





Publications
Presentations/Lectures
|
|
|



Nancy Gerits
A thesis submitted in partial
fulfilment
of the requirements for the degree of Philosophiae doctor
October
2007
Abstract
The protein kinase, MK5, belongs to the mitogen-activated protein kinase-activated protein kinases (MAPKAPK or MK), where it resembles MK2 and MK3 most. Despite the current knowledge about MK2, the kinases MK3 and MK5 remain poorly characterised. Therefore, we set out to determine in which cellular and physiological processes MK5 plays a role. Our studies included 1) the study of cAMP-dependent protein kinase (PKA) as an interaction partner for MK5; 2) a microarray analysis on the differential gene expression in the presence or absence of constitutive active MK5; 3) the study of transgenic mice that express a constitutive active variant of MK5; 4) the study of different signals that can activate the MK5 promoter.
First, we looked for new interaction partners for MK5. For this study, we analysed the sequence of MK5 for motifs that play a role in the PKA signalling pathway. We found four putative PKA binding sites and a partially conserved PKIrev signal. Subsequent in vitro kinase assays using PKAc® and MK5 demonstrated increased phosphorylation. In vivo kinase assays and co-immunoprecipitations confirmed the interaction and showed that PKA could phosphorylate and activate MK5. Since activation of the PKA pathway stimulates neuronal outgrowth and differentiation in PC12 cells, we checked whether the PKA signalling pathway could use MK5 to induce rearrangements of the actin cytoskeleton. Indeed, F-actin rearrangements occurred when active MK5 relocalised to the cytoplasm in response to activated and nuclear PKAc®.
Our second study involved the construction of a stable inducible cell line that expressed constitutive active MK5 upon the removal of doxycycline. We analysed the effect of this MK5 mutant on gene expression by microarray experiments. We found that genes involved in cytoskeletal processes, transcription and translation, signalling and metabolism were altered. These results could help to explain part of the phenotype of a C57BL/6 mouse line that expressed the same constitutive active MK5, which we constructed for our third study. Based on observations during background breeding, we performed two anxiety-related tests, the elevated plus maze (EPM) and the light-dark box (LD) test. These tests revealed unexpected gender differences in the behaviour of the mice on both tests: Female transgenic mice explored the open arm of the EPM longer than female non-transgenic mice, whereas male transgenic mice displayed increased locomotor activity in the LD test.
Finally, since upstream signals that elicit MK5 activity remain largely unidentified, we treated PC12 cells with different stimuli that converge to transcription factor binding sites in the MK5 promoter. This would allow us to assess which ones of those influence MK5 gene transcription. We found that only forskolin and heat shock could increase MK5 transcript levels and that cAMP-response element-binding protein (CREB) bound to the CRE motif in vitro. The same stimulus could not increase transcript levels in transient transfection studies with a reporter plasmid containing an 850 base-pair fragment of the human MK5 promoter encompassing the CRE motif. However, deletion of this CRE motif reduced the basal promoter activity about 10-fold.
Acknowledgements
First of all, I thank my supervisor Ugo for giving me the opportunity to research signalling pathways in his group. His expertise, support and patience have been invaluable to this work. Aside from being an excellent scientific adviser, both Ugo and Marijke also helped me to integrate in a foreign country, for which I am very grateful.
I also thank Mona for introducing me to a whole range of molecular biological techniques, for her patience and guidance. In addition, she was my main ``Norwegian'' teacher :-) Takk !
Also a big thanks to ``The Mk5 People'' : Sergiy, Alexey, Theresa, Helle and Olga for being such a happy, friendly and productive team! In particular, I would like to thank Olga and Helle for helping me out when I needed it most. Finally, a big hug for Marte and Conny for creating a happy atmosphere in the lab and beyond :-)
I appreciate the help from the ``mekanisk verksted'', for the construction of the elevated plus maze and the light dark box, and the animal facility for providing the mice with food and water, for separating them at weaning, for shifting their cages and for collecting blood samples. Also a kind word for the mice, for their courage and patience in undergoing all these elaborate tests in the hands of such a giant creature :-D
Thank you Marijke, Vigdis and Mona Nystad at Medical Genetics for the help and advise on southern blot, FISH and qPCR; Lotte and Halvor at Labforum for the help on the microarray experiment; Lars at the University Hospital for help with the histological slices; Randi, Helga-Marie and Tom for the small peek in the realm of electron microscopy and for storing the mouse tissues; Jill and Hans at the Norwegian Transgenic Centre (Oslo) for the microinjection; Mark Lensink at the Universitée Libre de Bruxelles (Belgium) for helping me explore the in silico world of proteins; and finally the people in Harwell (UK) and Munich (Germany) for their advice on how to work with transgenic mice.
A huge thanks goes to the group of Belgian friends and family, especially my parents for their support and Ingrid and Sara for all the peptalk and refreshing e-mails. Werner also deserves special thanks, for helping me out with the the collection and processing of large amounts of data, for the help on statistics and also for his realism, focus and support.
This research was funded through the Norwegian Research Council (NFR) from January 2004 until December 2007 and carried out at the Department for Microbiology and Virology, Section for Virology, at the University of Tromsø(Norway).
Contents
List of Abbreviations
aa : Amino Acid
AC : Adenylyl Cyclase
ARE : Adenylate/uridylate-Rich Elements
CA : Constitutive Active
CNS : Central Nervous System
DKO : Double knockout
DMBA : 7,12 Dimethylbenz(a)anthracene
ds : Double-stranded
EGF(R) : Epidermal Growth Factor (Receptor)
EGCG : Epigallocatechin gallate
(E)GFP : (Enhanced) Green Fluorescent Protein
EPM : Elevated Plus Maze
FGF(R) : Fibroblast-like Growth Factor (Receptor)
FSK : Forskolin
GF : Growth Factors (EGF, FGF, PDGF)
GPCR : G-Protein Coupled Receptor
Hsp : Heat-shock protein
KGF(R) : Keratinocyte Growth Factor (Receptor)
KI : Knock-In
KIM : Kinase Interaction Motif
KO : Knock-Out
LD : Light-Dark box
LPS : Lipopolysaccharide
LTD : Long Term Depression
LTP : Long Term Potentiation
MAP3K : MAPK Kinase Kinase, or MEKK
MAP2K : MAPK Kinase or MEK
MAPK : Mitogen Activated Protein Kinase
MBP : Myelin Basic Protein
MK : MAPK-Activated Protein Kinase or MAPKAPK
MKP : MAP Kinase Phosphatase
MNK : Mitogen Interacting Kinase
MSK : Mitogen and Stress-activated Protein Kinase
NES : Nuclear Export Signal
NLS : Nuclear Localisation Signal
NMDA : N-methyl-D-aspartate
NTG : Non-transgenic
NTM : Neurotransmitters
PDE : Phosphodiesterase
PDGF(R) : Platelet-Derived Growth Factor (Receptor)
PKA : Protein Kinase A
PKC : Protein Kinase C
PKI : Inhibitor of Protein Kinase A
PMA : Phorbol-12-Myristate-13-acetate = TPA
PRAK : P38-Regulated and Activated protein Kinase, also
MK5 or MAPKAPK5
res : Residues
RNAi : RNA interference
RNPII : RNA Polymerase II
RSK : Ribosomal S-Kinase, also p90^{RSK}
RTK : Receptor Tyrosine kinase
siRNA : Small Interfering RNA
shRNA : Short Hairpin RNA
ss : Single-Stranded
TF : Transcription Factor
TG : Transgenic
TSS : Transcriptional Start Site
WT : Wild-type
I. Introduction
1. Introduction into signalling cascades
Nothing lasts..
everything is changing into something else - T. McKenna
An essential way that guaranties the survival and normal functioning of the cell in a multi-cellular organism lies in its ability to properly communicate with other cells. This occurs by means of exchanging signals under the form of small molecules, like hormones and neurotransmitters. These signals bind specifically on their complementary targets, receptors, at the surface or on the inside of the cell, depending on the lipophilic nature of the signal. The surface receptors consist of an extracellular part that sticks out of the cell, a part that anchors the receptor to the cell membrane (a transmembrane domain) and an intracellular domain. Binding of a signal to its receptor causes a structural reshaping of the receptor, called a conformational change, that affects the intracellular domain as well. The alteration of the intracellular domain may uncover binding sites previously shielded from intracellular components and lead to the detachment of some components and the binding of others. These changes in turn trigger conformational changes in the partners bound to them and propagate throughout downstream components, affecting their function. This conversion of an extracellular signal into an intracellular one and its transmission intracellularly to elicit a response from the cell is called signal transduction. The end result of all these alterations depends on the nature of the signal and the functional status of the cell at the time the signal arrives. Translation of a signal into a cellular adaptation often requires protein kinases that transmit, amplify and convert the signal into a biological response (216,140).
In this dissertation, the activation and putative function of one enzyme - the MAPK Activating Protein Kinase 5, MK5 or PRAK- in the signal transduction pathway of the Mitogen Activated Protein Kinases will be discussed. However, in order to explain the complex world of proteins and their interactions with each other, some basic concepts will be introduced first.
1.1 Protein kinases
Within the world of signal transduction, the main actors are called protein kinases. They are enzymes that catalyse the transfer of the °-phosphate group of a trinucleotide (e.g. ATP) onto their substrate. This usually leads to a change in the activity or accessibility of the substrate. Since this action is reversible, protein kinases provide a fast way to cope with the ever-changing extracellular environment and enable a cell to react with speed to new stimuli. Protein phosphatases counteract the function of protein kinases by removing phosphate groups from their substrates.
The fact that approximately ~1.7% of the human genes encode for protein kinases, and aberrant action of those enzymes may contribute considerably to several diseases (cancer, immunoinflammatory diseases, infection) illustrates their importance. Protein kinases exist in both prokaryotes and eukaryotes, however, since this dissertation handles about eukaryotic organisms (mammalia) and focuses on MAPKs, only the family of the mammalian protein kinases and their MAPK members will be discussed.
1.1.1 The Eukaryotic Kinome
Sequence alignments of functional domains (domain phylogeny) reveals 7 major clusters within the eukaryotic protein kinase superfamily. Each cluster contains several subdivisions. Most eukaryotic organisms seem to encode members of all these clusters, except for the absence of Tyrosine Kinases (TK) and Tyrosine Kinase Like kinases (TKL's) in worm. The human genome seems to encode 518 protein kinases for which the overview below describes the different clusters and subdivisions that comprise MAPKs (see also Figure 1.1) (132,234).
|
|
||
|
|
|
|
|
|
||
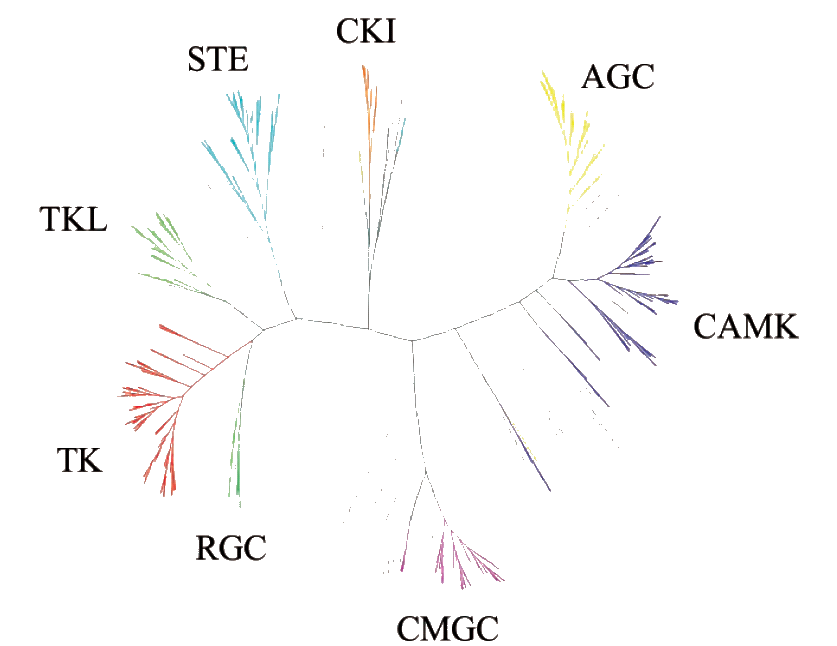
Figure 1.1: Phylogenetic tree of the different protein kinase families (234).
The oldest clusters comprise :
-
The Tyrosine Kinase cluster (TK) : it consists of approximately 84 members divided over 25 families.
-
The AGC cluster : named after the PKA, PKG and PKC families. It contains 61 cyclic nucleotide- and calcium-phospholipid-dependent kinases divided over 13 families. This cluster contains the MAPKs : the Ribosomal-S6-kinases RSK1, RSK2, RSK3 and RSK4 and the Mitogen and Stress Activated Kinases MSK1 and MSK2.
-
The Calmodulin-dependent kinase cluster (CaMKs) : consists of 66 members in 15 families of which five kinases signal in the MAPK pathway : MAPKAPK2, MAPKAPK3, MAPKAPK5, MNK1 and MNK2.
-
The CDK-MAPK-GSK-CLK cluster (CMGC): comprises 61 members divided into 8 families. The MAPKs ERK1, ERK2, ERK3, ERK4, ERK5, ERK7, JNK1, JNK2, JNK3, NLK, p38®, p38¯, p38° , and p38± all belong to this cluster.
A more extensive study based on phylogenetic analysis in C. elegans and Drosophila led to the addition of three more clusters:
-
The STE cluster contains kinases that function in the MAPK cascades and is based on the analysis of sterile yeast mutants. They contain 45 members in 4 families. For example the MAP2Ks (1 to 7) and the MAP3Ks (1 to 8) belong to the STE 7 and STE 11 family respectively. Other MAP3Ks like Cot and NIK belong to the STE-Unique family within the STE cluster.
-
The CK1 cluster, named after Casein Kinase 1, is greatly expanded in worm. It consists of 11 members in 3 families.
-
The Tyrosine-Kinase Like (TKL) kinases include the STKR family of TGF-¯ serine/threonine kinase receptors and as the name suggests is phylogenetically closely linked to the TK's. This cluster contains 37 members in 7 families. For example, MAP3Ks Raf (Raf family), Mos (Unspecified division), MLK/TAK (MLK family).
There exists another cluster with atypical kinases. This cluster consists of 63 members in 24 families.
1.1.2 Physiological relevance
Obviously, protein phosphatases and kinases should function appropriately in order for signalling pathways to run smoothly. Deregulated protein kinases often play an important role in disease development and progression, therefore, researchers search for molecules that can inhibit ill-tuned kinases. These protein kinase inhibitors can mimic ATP sites of a protein kinase or -less common- bind its substrate. They may bind a receptor and prevent downstream signalling or induce the RNA interference machinery when the inhibitor is expressed as siRNA or shRNA. Protein kinase inhibitors against a specific class of kinases, e.g. MAPK inhibitors, allow the functional study of MAPK and in addition provide an important means for the development of therapeutics against diseases (see Table 2.1).
For example, activating B-Raf mutations occur in 30-60% of the melanomas, 30-50% of thyroid cancer cases and 5-20% of the colorectal cancers. The anti-sense inhibitor LErafAON and the small molecule Sorafenib can both inhibit B-Raf activity and inhibit further progression of these cancers (122,243). Other examples of protein kinase inhibitors are carboline analogues that bind in a region near the p-loop and the hinge region in MK2. These compounds also display moderate selectivity against MK1/RSK and ERK2 (404). Another set of inhibitors against MK2 comprises pyrrolopyridine inhibitors. These inhibitors suppress MK2-induced tumour necrosis factor (TNF) production in a cellular model of inflammation and may become promising candidates for the treatment of rheumatoid arthritis and other inflammatory diseases (7). For the MK2-related kinase MK5, there exists currently only an aspecific inhibitor, epigallocatechin gallate (EGCG), extracted from green tea. This natural polyphenolic compound reduces the activity of DYRK1A and PRAK by 50% when administered in 0.33 ¹M and 1 ¹M concentrations respectively (20).
1.2 Initiation of signalling cascades
As mentioned in section 1, an extracellular signal binds to its receptor, which then undergoes a conformationals change. There exist several types of receptors : ion channel receptors, G-protein coupled receptors (GPCR), tyrosine kinase receptors (RTK) and intracellular receptors. The nature of the signal determines which type of receptor becomes activated. MAPKs become activated by stress signals (like UV and osmotic stress) and growth factors (GF) e.g. EGF, PDGF and FGF. These GF bind to their respective receptors (EGFR, PDGFR, FGFR) on the cells' surface. Ligand binding leads to dimerisation and subsequent autophosphorylation of the conserved tyrosine residues on the intracellular part of the receptor. The phosphorylated residues then provide a binding site for the conserved Arg within the Src-homology domains (or SH2 domains) of linker proteins like e.g. Shc (see 1.2.1). These molecules that connect the receptor to the downstream signalling cascade are called linkers and adaptors.
1.2.1 Linkers and Adaptors
The adaptor Shc possesses an Src homology-2 (SH2) domain, which can recognise a phosphorylated tyrosine on an activated receptor. Their interaction leads to the phosphorylation of the Tyr within Shc, which subsequently serves as a marker for other proteins that contain SH2 domains e.g. Growth factor Receptor Binding 2 or Grb2. When the SH2 domain of Grb2 binds to Shc, the two Src-homology 3, or SH3, domains remain available for binding to Pro-rich regions of two other proteins. Son Of Sevenless (SOS) and GTPase-Activating Proteins (GAP) are examples of proteins that can simultaneously bind to Grb2. The former exchanges GDP for GTP, whereas the latter promotes the reversed exchange. This GDP/GTP exchange cycle regulates the activity of the monomeric G-protein Ras. Activation of Ras leads to the activation of its GTPase activity and results in the activation of a MAP3K (Raf) and subsequent activation of the MAP kinase cascade (see chapter 2) (140).
1.2.2 Monomeric G-proteins
The 21kDa Ras functions as a cellular proto-oncogene, that is related to the RAt Sarcoma retro-viral oncogene. It receives lipid moieties like prenyl and farnesyl groups to become anchored to the membrane. There it can interact with linkers that transmit signals from activated receptors to Guanine nucleotide Exchange Factors (GEFs) and GAPs, which regulate Ras' activity. Ras requires these exchange factors because it lacks the vital guanidino group in Arg that becomes partially negatively charged during phosphoryl transfer and which activates the kinase. Besides activating the MAPK Raf, Ras can also activate Pyk and PI3K (140).
Other monomeric G proteins are Rap, Ran and Rho/Rac/cdc42. The former regulates vesicular protein traffic and requires e.g. Epac as GDP/GTP exchanger (see section 3.5). Ran mediates transport in and out of the nucleus and in its activated form, it can induce microtubule re-organisation and formation of the mitotic spindle in Xenopus eggs. Rho/Rac/cdc42 belong to a subgroup of the Ras superfamily and consist of Rho A, B, C, G, Rac1 and Rac2 and cdc42. They mainly activate the p21-Activated Kinases (PAKs) and their downstream components, the JNKs (see section 2.2.3), which regulate the cytoskeleton and the cell cycle (140).
2. The MAPK signalling pathway
The MAPK signalling pathway consists of individual components, called MAP kinases. MAP Kinase stands for Mitogen Activated Protein Kinase. As the name suggests, mitogens were identified as the first group of signals that stimulated this pathway. Other signals that elicited MAPK activation soon joined the list e.g. environmental stress factors like UV irradiation, but also cytokines involved in inflammation. Activation of the MAPK signalling pathway comprises four levels of regulation by phosphorylation : the first, where a MAP kinase kinase kinase (also MAPKKK, MAP3K or MEKK) becomes activated by transfer of a phosphate group onto the serine or threonine residue within its activation loop (72). This kinase then phosphorylates the serine residue within the activation loop of the MAP kinase kinase (MAPKK, MAP2K or MEK), thereby activating the kinase. These kinases are also called dual specificity kinases because they phosphorylate and activate their substrate, a MAP kinase or MAPK, on the two conserved residues, in the motif of the activation loop (356). Their activation ultimately results in the transfer of a phosphate group onto the serine or threonine residues of non-kinase substrates (e.g. transcription factors) or other kinases, like the MAP Kinase Activated Protein Kinases (MAPKAPK or MK). Figure 2.1 illustrates the basic components of the MAP kinase signalling module, although the in vivo situation is somewhat more complex.
|
|
||
|
|
|
|
|
|
||
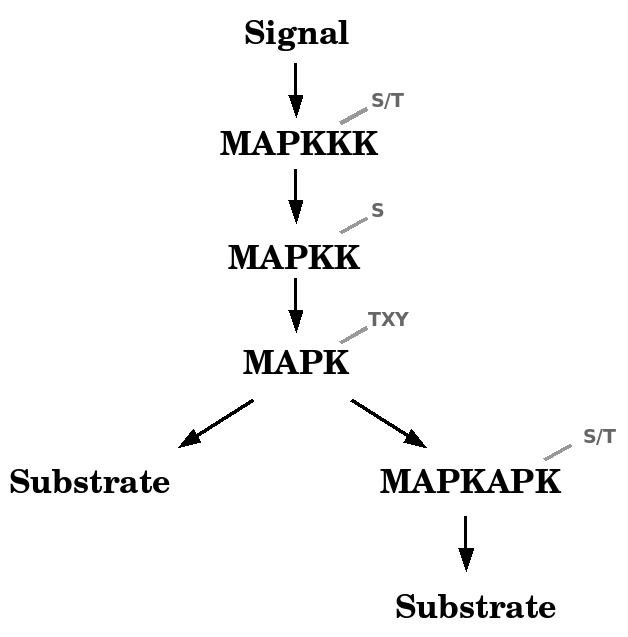
Figure 2.1: The basic MAPK module. A MAP3K becomes activated by a phosphate group transfer from upstream linkers and adaptors with kinase activity. A phosphorylated MAP3K in turn phosphorylates and activates a MAP2K, which activates the downstream MAPK. MAPKs then phosphorylate their substrates or yet other MAPKs, the MAPKAPKs, which then phosphorylate their substrates. The grey superscribed letters indicate which amino acids will receive the phosphate group (phosphoacceptor site).
There exist several subdivisions of MAPK pathways based on the different types of MAP kinases that become activated. In section 2.2 and accompanying Figure 2.3, each branch of the MAPK signalling pathway in terms of their sequence, structure and activating signals will be discussed. For now, it suffices to say that particular stimuli activate particular branches of the MAPK pathway and that each branch is named after the particular MAPK that becomes activated in this pathway:
-
Mitogens or growth factor signals activate the classical MAPK pathway or the ERK1/2 pathway discussed in section 2.2.1.
-
Mitogens, growth factor signals and stress factors can activate the ERK5 pathway (see section 2.2.2).
-
Stress factors activate the JNK pathway and the p38 pathway. Sections 2.2.3 and 2.2.4, respectively, reveal more about these pathways.
-
The upstream signals of most of the atypical MAPKs remain unidentified (see section 2.2.5 for more information).
2.1 Structure of MAPKs
2.1.1 Features of MAPK structure
The primary sequence of MAP kinases, and protein kinases in general, reveals particular conservation of 11 regions, indicated by Roman numbers (132). When the kinase folds, these conserved domains form a common bilobal structure that consists of an N-terminal and a C-terminal lobe connected by a hinge region. The cleft between the N- and C-terminal lobes forms the catalytic site. In unphosphorylated MAPKs an inhibitory ®-helix prevents access to the site, whereas phosphorylation catalysed by an upstream kinase removes the ®-helix, opens the catalytic cleft and activates the kinase (92,359).
The N-terminal lobe of a MAPK screens for extracellular signals, like ATP, and binds them (132). The conserved Lys in subdomain II and the conserved Asp in subdomain VII serve to anchor and orient the ATP, and thereby facilitate the transfer of its °-phosphate group. In addition, the N-terminal lobe contains a loop, the L16-loop or phosphorylation lip, in proximity of the catalytic cleft. Within this loop resides the phosphorylation motif, TXY, of the MAPK which becomes phosphorylated by the upstream MAP2K.
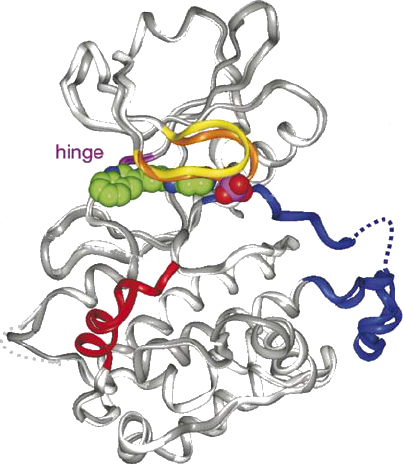
Figure 2.2 illustrates some of these structural features in the crystal structure of MAPKAPK2 (see section 2.3.4) as an example. MAPKAPK2 consists of a Pro-rich N-terminal domain (res.10-44) which interacts with SH3 domain-containing proteins, a kinase domain (res.51-325) with a conserved Thr in the phosphorylation loop and regulatory phosphoacceptor residues in the hinge region. The auto-inhibitory helix (res.328-364) in the C-terminal domain contains a NES and bipartite NLS signalling sequence (res.356-368 and res.373-389 respectively), where the latter can bind p38 (143). The MAPKAPK2 ATP binding site lies in the cleft between the N-terminal and C-terminal lobe and consists of a roof formed by the Gly-rich nucleotide binding loop (res.71-76) and a floor formed by the catalytic loop (res.184-193) (143).
|
|
||
|
|
 |
|
|
Figure 2.2: The superimposed structure of two MK2 crystals with the specific inhibitor, compound-1 bound in the ATP site. The crystals are depicted in dark grey (form1) and light grey (form2). The inhibitor is shown in CPK representation. The N-terminal lobe consists mainly of a conserved ®-helix and a 5-stranded ¯-sheet. The C-terminal consists predominantly of ®-helices. The high affinity inhibitor, compound-1, binds deeply inside the ATP pocket guided by H-bonds and Van Der Waals bonds. Colours : nucleotide binding loop in yellow (form1) and orange (form2), hinge region in magenta, activation segment in blue (with the disordered part in dots) and the auto-inhibitory domain in red. The figure is adapted from (143). |
||
2.1.2 Substrate binding domain
The C-terminal domain binds the substrate of the MAPK. Since each MAPK recognises a selective range of targets, the region for interaction between both proteins must consist of residues specific for the interaction. In contrast to other protein kinases like PKA, MAPKs lack specific consensus sites for interaction. Therefore, MAPKs carry docking domains, which lie opposite of the active site. The first docking domain, termed conserved common docking domain or CD domain, contains acidic and hydrophobic residues. The second domain, the ED domain, consists of an acidic Glu (E) and Asp (D) and conveys docking specificity to the MAPK. For example, substitution of the ED site in ERK2 (Thr157 and Thr158) by the ED site of p38 (Glu160 and Asp161) converted the docking specificity of MK3 from p38 to the mutated ERK2 (307,359).
Since the CD and ED domains contain acidic residues, they serve to interact electrostatically with the D-domains on the MAPK substrates, which are basic in nature. These docking domains received their name after the first domain discovered: the ±-domain of c-Jun. Several MAPK substrates and regulatory proteins e.g. MAPKK, MKPs, which dephosphorylate MAPKs, and Kinase Suppressor of Ras (KSR) possess such domains. In addition to its basic nature, the D-domains also carry some hydrophobic residues (an LXL motif and/or a triplet of hydrophobic residues) which can interact with the hydrophobic residues near the CD domain on the MAPK via hydrophobic interactions. Besides D-domains, MAPK substrates can also contain a DEF domain, named after ``Docking site for ERK and FXFP''. This domain, discovered in C. elegans, consists of FXFP residues, and is required for efficient substrate phosphorylation. For example, Elk1 and KSR possess both a D domain and a DEF domain. The presence or absence of these features provides a 3D environment for the interaction between a MAPK and a specific substrate (307,23).
2.2 MAP kinase signalling pathways
In order to elicit MAPK activation, MAP2Ks phosphorylate both the Thr and Tyr of the T-X-Y (Thr-X-Tyr) motif within the activation loop. Activated MAPKs target Ser-Pro and Thr-Pro residues on their substrates and may translocate to another cellular compartment upon activation: inactive MAPKs reside predominantly in the cytoplasm, whereas activated MAPKs usually translocate to the cell nucleus (264,75). MAPKs also deploy scaffold proteins in order to allow fine tuning of the pathway. These proteins serve to bring together the MAP3Ks, MAP2Ks, MAPKs and their substrates, so as to allow them to interact specifically. First, it was thought that scaffolds were static entities, only providing docking sites to MAPKs for their interactions. However, lately, it seems scaffolds can also contribute actively to a particular signalling cascade (413,110,49,78,250).
This section describes the different MAP kinase pathways in terms of their activation signals, components and relevance. Figure 2.3 summarises these different MAPK pathways and their components.
|
|
||
|
|
|
|
|
|
||
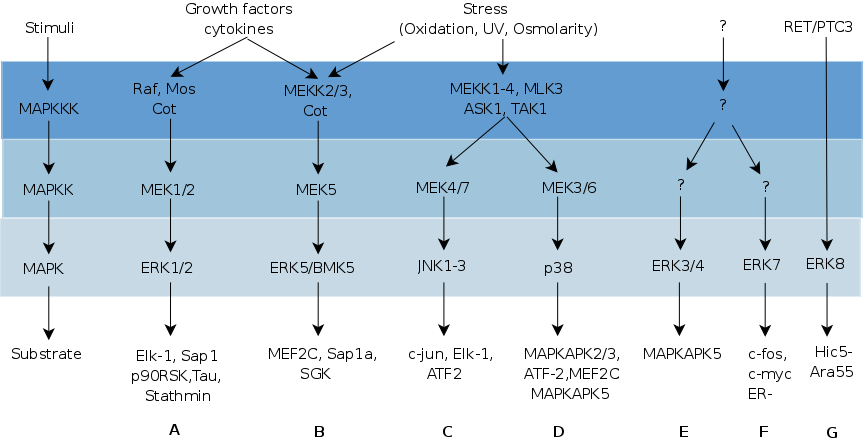
Figure 2.3: The different MAPK pathways with their upstream activators, MAPK components and downstream targets. For more details, see section 2.2 and subdivisions. The figure is adapted from (167).
2.2.1 ERK or Extracellular signal-Related
Kinases
The ERK pathway (Fig 2.3A), also known as the classical MAP kinase signalling pathway, consists of the MAPKKKs A-Raf, B-Raf, and c-Raf-1, the MAPKKs MEK1 and MEK2, the MAPKs ERK1 and ERK2. Cytokines, growth factors, serum, certain stresses, ligands for G protein-coupled receptors and microtubule disorganisation can all activate A-Raf, B-Raf and c-Raf-1 in a Ras-dependent way. Those kinases then differentially regulate the activity of their downstream effectors MEK1 and MEK2 by phosphorylation. These highly homologous isoforms phosphorylate ERK1 and ERK2 (307).
ERK1 and ERK2, respectively 44 and 42 kDa in size, share 90% sequence identity and their ubiquitous expression pattern (37). The phosphoacceptor site of ERK1 and ERK2 consists of a Thr-Glu-Tyr (TEY) motif. Activated ERK1 and ERK2 can target transcription factors (e.g. Elk1, Sap1, STAT), cytoplasmic proteins (e.g. phospholipase A2) and the kinases MNK1, MNK2, MSK1, MSK2, RSK1/2/3, p70S6K and p70S5K. These substrates subsequently influence cellular processes like proliferation, differentiation and survival (120,307). For example, failure of ERK1/2 to translocate to the nucleus prevents activation of Elk-1 and blocks the ability of cells to initiate DNA synthesis in response to GF (140,120).
Some scaffold proteins support the interaction of components in the ERK pathway. For example, KSR gathers the Raf/MEK/ERK module together. Other scaffold proteins that function in the ERK pathway comprise : MP1, MEKK1, ¯-arrestin1/2 [see 78 and references therein].
2.2.2 ERK5 or BMK
Big MAPK1, BMK1 or ERK5 (Fig 2.3B), earned its name from its size (98kDa), being approximately twice as big as ERK1 and ERK2. Except for the liver, all tissues express ERK5. It shares approximately 66% sequence similarity with the kinase domain of ERK1/2 and shares their TEY phosphorylation motif. There exist several reports that indicate the existence of three splice variants, with the possibility for two of them to act in a dominant-negative fashion to fine tune the MEK5/ERK5 pathway (422). The ERK5 pathway becomes activated both by mitogenic stimuli like ERK1 and ERK2, as by oxidative stress like JNK and p38. These stimuli may transmit their signal through MEKK2 and MEKK3 to MEK5 and ERK5 (217).
ERK5 also possesses an extensive C-terminal tail of unknown function that contains several Pro-rich motifs. These motifs can allow for interaction with SH3 domain-containing proteins (209). Downstream substrates of ERK5 include transcription factors such as c-Myc, NF-·B, Sap1a, c-Fos, Fra-1 and MEF2 family members, and the serum- and glucocorticoid-inducible kinase (37). These downstream targets connect ERK5 to physiological processes like the maintenance of vascular integrity, cellular proliferation, differentiation and survival (408,209).
2.2.3 JNK or Jun N-terminal kinases
The JNKs (Fig 2.3C) were originally discovered as p54-microtubule-associated protein kinases that were activated by cycloheximide. In addition, they phosphorylated the NH2-terminal part of the c-Jun transcription factor. Similar to the p38 pathway (see 2.2.4), the JNK pathway becomes activated by stress and UV stimuli, hence the name stress activated protein kinases 1 or SAPK1. These stimuli transmit their signals through Rho family receptors, tyrosine kinase receptors and cytokine receptors to MAP3Ks MLKs, ALK, TAK, similar to the p38 pathway. However, activation of the JNK pathway occurs specifically via MAP2K MKK4 and MKK7. MKK4 and MKK7 reside in the cytoplasm and nucleus, but they respond differently to incoming stimuli: pathogenic virulence factors can activate both MKK4 and MKK7, whereas environmental stress mainly activates MKK4 and cytokines activate MKK7 (84). There exist several reports of MAPKs upstream of MAP3Ks, the MAP4Ks 1 to 6, which all specifically activate the JNK pathway (392).
Activation of the JNKs occurs through phosphorylation of the TPY motif in the activation loop. The difference between which JNK becomes activated, depends on the tissue expression: most tissues express JNK1 and JNK2, whereas JNK3 expression is mainly restricted to the brain, heart and testis. In addition, each JNK can be expressed as different splice forms, as 46kDa or 55kDa proteins depending on the absence or presence of a C-terminal tail whose function is unknown (84). The physiological significance of the JNK pathway lies in apoptotic and survival pathways, embryogenic morphogenesis, tumour biology and immunological diseases (167,84).
Different scaffold proteins regulate the interaction between different components of the JNK pathway. MEKK1, JIP1/2/3, JLP(=JIP4), MKPX, POSH, IKAP, CrkII, ¯-arrestin2 and SKRP1 all bind components of the JNK pathway [78,250 and references therein].
2.2.4 p38
The discovery of the p38 (Fig 2.3D) pathway started when stimulation of macrophages with lipopolysaccharide (LPS) led to the tyrosine phosphorylation of a protein of 38kDa, p38® , and its subsequent characterization as a specific target of pyridinyl imidazoles. The p38 MAPKs or Stress Activated Protein Kinases 2 (SAPK2) consist of p38® (SAPK2a), p38¯ (SAPK2b), p38° (SAPK3) and p38± (SAPK4) and are activated by phosphorylation of the TGY motif within the activation loop. Upstream activators of the p38 pathway comprise stress stimuli like UV radiation, sodium arsenite, heat shock, bacterial LPS, and pro-inflammatory cytokines (324). These stimuli caused activation of the MAP2Ks MKK6 and MKK3. Whereas MKK6 can activate all four kinases, MKK3 activates all but p38¯ (324). Phosphorylation of the conserved residues in the activation loop results in a conformational change which turns the active site into a site with high catalytic activity. Similar to the JNK pathway, some MAP4Ks (MAP4K2 and MAP4K6) can regulate the activation of the p38 MAPK pathway (392) and scaffold proteins can assist in the regulation of this pathway e.g. OSM, JIP2 and JIP4 [see 78 and references therein].
p38® and p38¯ share 74% amino acid sequence homology, their wide expression pattern and their inhibition by the pyridinyl imidazole SB203580. p38° and p38± share respectively 63% and 61% of their amino acid sequence with p38® . Both kinases have a more limited expression pattern: the former being mainly expressed in skeletal muscle, whereas the latter can be found predominantly in testis, pancreas, small intestine, and CD4+ T-cells. Additionally, p38 °, also called ERK6, displays a distinctive subcellular localisation pattern and its interaction with PDZ domain-containing proteins set it apart from the other p38 MAPKs (37). Aside from their high sequence identity, p38 MAPKs also share wide substrate similarity, although the activity towards these same substrates can differ substantially. The very diverse p38 pathway plays roles in regulating the immune systems response, mammalian preimplantation development, cell survival and death, differentiation and growth (167,391,120,307,13).
2.2.5 The atypical MAPKs ERK3, 4, 7 and 8
Besides the well-characterised MAPK pathways, there exist several MAPKs whose upstream activators and downstream targets remain poorly identified. These MAPKs, called the atypical MAPKs, comprise ERK3, ERK4, ERK7 and ERK8.
ERK3 (MAPK6)
ERK3 was originally isolated as an ERK1 homologue. Sequence comparison between ERK1 and ERK3 reveals that they share 83% and 72% homology in kinase domains V and IX. Aside from this similarity, ERK3 differs from the conventional ERKs in its unique C-terminal extension, its APE domain which is replaced by the SPR domain, and a different phosphorylation motif (37). The phosphorylation motif in ERK3 exists as a SEG motif (in stead of a TEY in other ERKs) of which the Ser189 is constitutively phosphorylated. This implies that phosphorylation events do not serve to regulate ERK3's activity, but that its regulation occurs by another means like its cellular abundance. For example, ERK3 expression increases during mouse development (E11) and during differentiation towards neuronal or muscle lineages in P19 embryonal carcinoma cells (42,379,325).
No upstream activators and hardly any in vivo downstream targets have been identified for ERK3. One interaction partner for ERK3, both in vitro and in vivo, is MK5 (320,325) (see 2.4). Recently Kant et al proved that ERK3 can also interact with ERK4 to form protein complexes, whereby the kinase activity of ERK4 is required for the activation of MK5 (187). Increased expression of PKC¯, which blocks differentiation of colon cancer cells, can also activate ERK3, although further studies are required to pinpoint the significance of this interaction (316). ERK3 also interacts with cyclin D3 and MAP2 (9,350). In vitro substrates for ERK3 include Myelin Basic Protein (MBP) and Histone H1 (320).
The N-terminal lobe of ERK3 contains 2 destabilisation regions which explains its very short half-life. ERK3 stabilisation and accumulation results in G1 cell cycle arrest and may depend on the stabilising effect of MK5 on ERK3 and the redistribution of ERK3 to the cytoplasm (184,320). Although there exists no proof as to the exact mechanism. Even more, ERK3 contains several motifs that resemble Leu-rich NES signals for Crm1/exportin-mediated nuclear export. However, which of those motifs play a role in ERK3s nuclear export remains to be determined (184). In addition, there exists some confusion about the subcellular location of ERK3 in untreated cells. A 62kDa truncated ERK3, with a different C-terminal tail resides predominantly in the nucleus, whereas ERK3 expressed by a plasmid containing human cDNA for ERK3, resides both in the nucleus and the cytoplasm (184,320,325).
The 400aa C-terminal tail of ERK3 shares some similarity with ERK4. It possesses 2 CD-like motifs for interaction with the basic D-domains in MAPK substrates (as identified in MKs), but these motifs differ from ERK1/2, JNKs and p38 MAPKs and their exact role remains enigmatic (320). Finally, the physiological role for ERK3 remains unestablished, although there exist some hints that it plays a role during G1 cell cycle arrest and during development, since ERK3 KO mice display embryonic lethality [unpublished results Meloche].
ERK4 (p63MAPK)
ERK4, also designated p63MAPK, remains a mystery as well. Similar to ERK3, its upstream activators and downstream substrates remain largely unidentified. Even more, the fact that ERK4 does not respond to different MAPK stimuli, only complicates further identification of interaction partners (37). Currently, only MK5 and ERK3 can interact with ERK4 in vivo, but no biological function has been found for ERK4 yet and no KO model for ERK4 has been constructed (426,320,187) (see 2.4). ERK4 displays 73% homology towards ERK3 and shares its SEG phosphorylation motif (187). Its tissue expression occurs mainly in the heart, brain and lungs (125).
ERK7
The amino acid sequence of the kinase domain of ERK7, a 61 kDa protein, shares ~40% homology to ERK1 and ERK2. Since neither MEK1 nor MEK2 phosphorylate ERK7, the upstream activators of ERK7 remain unknown. Like ERK3, ERK7 seems to be constitutive active due to autophosphorylation. Downstream substrates of ERK7 include the transcription factors c-Fos and c-Myc and the estrogen receptor-® (ER-® ). ERK7 also interacts directly with the intracellular chloride channel CLIC3, but does not phosphorylate it (37). The 547aa protein contains a TEY motif like other ERKs and seems mainly present in the nucleus where it may suppress DNA synthesis as a part of its anticipated cytostatic or cytotoxic role (37).
ERK8
ERK8 contains 544aa and shares an overall amino acid identity of 69% (82% in the kinase domain) with ERK7. This 60kDa protein becomes activated by serum in a Src-dependent way [reviewed in 37]. Recently, the constitutive active form of RET, RET/PTC3, could activate ERK8 and this led to a direct interaction and regulation of Hic-5/ARA55 activity, a LIM protein that functions as a cofactor for nuclear receptors (160,312). Its slow activation implies a role in long term reponses to mitogenic factors (37).
2.3 The MAPK-activated protein kinases
As the name implies, the MAPK-activated protein kinases, MAPKAPKs or MKs for short, constitute a group of kinases targeted by MAPKs. Similar to MAPKs, they become activated by the transfer of a phosphate group onto their Ser/Thr motif. Their activation leads to the phosphorylation of downstream targets like small heat shock proteins (Hsp) and transcription factors (TF) like cAMP-response element-binding protein (CREB), serum response factor and basic helix-loop-helix protein E47.
|
|
||
|
|
|
|
|
|
||
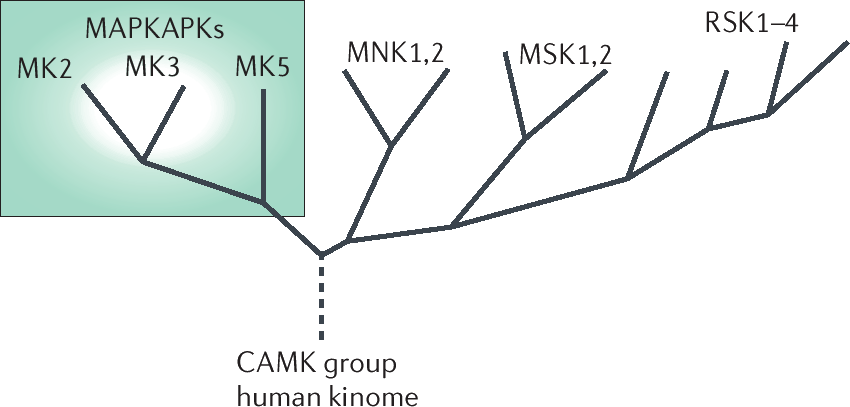
Figure 2.4: Phylogenetic tree from MAPKAPKs. The MAPKAPKs belong to the CaMK kinases and consist of the ribosomal-S-kinases, the mitogen and stress-activated protein kinases, the mitogen-interacting kinases and the MKs consisting of either MK2 and MK3 or MK5. The text discusses each of these branches (117).
Figure 2.4 indicates the different branches of the phylogenetic tree of MAPKAPKs : the Ribosomal-S-Kinases (RSK), the Mitogen and Stress-activated protein Kinases (MSK), the Mitogen-interacting Kinases (MNK) and the MKs consisting of either MK2 and MK3 or MK5. This section discusses each of these branches in terms of their structure, their activation, their substrates and their physiological relevance. These characteristics are also summarised in Table 2.1.
2.3.1 Ribosomal S6 kinases (RSK)
The 90kDa ribosomal S6 kinases, RSKs for short, were named after kinases that could phosphorylate the S6 protein, a 31kDa component of the 40S ribosomal subunit in vitro (420). Although their name suggests the contrary, they do not phosphorylate this protein in vivo. The RSKs consist of four members: RSK1 (MAPKAPK1a), RSK2 (MAPKAPK1b), RSK3 (MAPKAPK1c) and RSK4 (MAPKAPK1d) (307). They share 73% to 80% sequence identity but are differentially expressed : RSK1 expression occurs in the CNS, adipocytes, the heart, the kidney, the liver, the lung, pancreas, placenta and skeletal muscle. Subcellularly, RSK1 resides in the cytoplasm, the nucleus and near membrane fractions (279,136,273). The CNS, skeletal muscle, fat, spleen, lungs, skeleton and kidneys all express RSK2. In addition, RSK2 appears also during murine development (96,367,199,103,282). RSK3 expression occurs in liver, muscle, placenta and T-cells. Subcellularly, it resides in the cytoplasm and nucleus. Abundant expression of RSK4 takes place in the brain, heart, kidney, liver, lung, pancreas, pituitary gland, skeletal muscle, thyroid gland and during murine development (412,99,279). As the last discovered RSK, the 746aa RSK4 differs from RSK1, RSK2 and RSK3 in that its serine residues are constitutively phosphorylated. This makes RSK4 not responsive to GF-induced stimuli (99).
2.3.1.1 RSK structure and activation
RSKs posses a two-kinase domain structure. The N-terminal kinase domain, referred to as NTK, resembles the kinase domain of the AGC group of kinases most (see 1.1.1). The C-terminal kinase domain, referred to as CTK, contains a MAPK binding site (indicated by a Leu-X-X-Arg-Arg motif) and is conserved in RSK1, RSK2 and RSK3 (307,333,273). This domain resembles the active site of phosphorylase b kinase and Ca2+/Calmodulin kinase (32). A hinge region that contains a hydrophobic motif, connects the two kinase domains. The appearance of two kinase domains implies a functionality for them. Indeed, full kinase activity (like acute NGF treatment or UV light (307,333,241)) requires the phosphorylation of key residues on both domains. In rat RSK1, the upstream kinases ERK1 and ERK2 phosphorylate Thr573 in the CTK and Thr359 and Ser363 in the linker region. Phosphorylation of these residues promotes the autophosphorylation activity of the CTK domain towards Ser380 in the linker region. This then creates a binding site for PDK1, which phosphorylates the Ser221 in the T-loop of the NTK. This modification decreases the affinity of RSK for ERK and leads to full activation of the RSK (99,333). The RSK subsequently phosphorylates downstream substrates like CREB, C/EBP¯ , Fos, Myt1, Bub1, BAD and 14-3-3 proteins (333). Dephosphorylation by protein phosphatase 2A (PP2A) leads to deactivation of the RSK (32).
Table 2.1: Summary of the substrates and biological functions of MAPKAPKs.
|
MK |
Substrate |
Function |
Disease |
Inhibitor |
|
RSK1 |
CREB, Fos, C/EBPβ, Myt1, Bub1, 14-3-3 |
membrane trafficking neuronal differentiation |
Rubinstein-Tabi syndrome, AD? |
BI-D1870, SL0101, fmk-pa |
|
RKS2 |
||||
|
Brain development osteoblast differentiation and function differentiation into muscle fibers |
Coffin-Lowry syndrome, prostate cancer? |
|||
|
RSK3 |
||||
|
RSK4 |
||||
|
? |
? |
SL0101 |
||
|
cell cycle, glucose metabolism? |
Mental retardation X-linked deafness type3 |
SL0101, fmk-pa |
||
|
MSK1 |
CREB, ER81, NF·B, histone H3, HMG14, tau |
neuronal cell death enhances transcription fear conditioning drug-induced locomoter sensitivity, drug-induced place preference |
? |
? |
|
MSK2 |
histone H3 |
? |
? |
? |
|
MNK1 |
eIF4E,Spry2 hnRNPA1,PLS2 |
protein synthesis stress responses, cytokine production |
? |
CGP57380 |
|
MNK2 |
eIF4E, ER® |
protein synthesis |
? |
CGP57380 |
|
MK2 |
TH, Hsp27, 14-3-3, LIMK1, cdc25, E47, TSC2, CREB, ATF1, hnRNP40A0 |
stress responses cell cycle cytokine production |
psoriasis, arthritis pancreatitis ischemia, AD?, atherosclerosis |
Compound1 Pps, carboline analogs |
|
MK3 |
E47, Bmi |
stress responses cell cycle cytokine production, regulation of gene expression |
? |
? |
|
MK5 |
ERK3/4, PKA, p53, 14-3-3ε, Hsp27 |
tumour suppression cell migration |
? |
? |
|
|
||||
2.3.1.2 Function of RSKs
The RSKs have been extensively studied in cell cultures and mouse models. In general, they play a role in cellular reponses to stress, in survival and proliferation of cells, in T-cell and platelet activation. They become activated by oncogenic transformation, stimulate the G0/G1 transition and phosphorylate nuclear substrates, protein phosphatase 1 (PP1) and glycogen synthase kinase 3 (32).
-
Specific RSK1 functions include regulation of membrane trafficking (136), neuronal differentiation of PC12 cells (333) and protection of lung epithelium via interaction with KGFR (273).
-
RSK2 plays an important role in the normal development of the brain (333), in the regulation of adipose mass and oocyte maturation in mice (170,103). RSK2 KO mice serve as a model for Coffin-Lowry syndrome. These studies showed that RSK2 is essential in osteoblast differentiation and function, and that deletion of RSK2 led to deficiencies in several types of memory (282). Furthermore, vaccinia virus and HIV both target RSK2 for their replication (142,8). RSK2 also affects differentiation into muscle fibers (57) and osteosarcoma development (81).
-
RSK4 affects the cell cycle (99) and seems (partially) absent in mental retardation or X-linked deafness type 3 (412).
2.3.2 Mitogen- and Stress activated protein Kinases (MSK)
Mitogen- and stress activated protein kinases (MSKs) comprise MSK1 (RLPK) and MSK2 (RSK-B). These kinases share 75% sequence identity with each other. They also possess the two-kinase domain as the one described in RSKs. Hence, their activation mechanism resembles that of RSKs, except for two differences. The first distinction lies in the MAPK docking site, where both ERK and p38 can bind. This means that MSKs can be activated by mitogenic stimuli as well as stress, in contrast to RSKs which are mostly activated by mitogenic stimuli. The second difference comprises the activation loop, which does not become phosphorylated by PDK1, but receives its phosphate group via autophosphorylation (99,340).
2.3.2.1 MSK activation
MSK1 requires phosphorylation of Ser360 and Thr581 by ERK1/2 and p38 for its activation. However, in order to become fully active, the CTK domain auto-phosphorylates Ser212 in the N-terminal loop and Ser376 in the hydrophobic motif. Mutation of another phosphoacceptor site, Ser381, reduces MSK1 activity by decreasing the phosphorylation of Ser376 and -to a lesser extent- that of Ser212. This might indicate that Ser381 plays a stabilising role during MSK activation. The meaning of the phosphorylation of Ser residues 750, 752 and 758 by the NTK remains elusive (238). In the inactive conformation, residues in the hydrophobic pocket which normally contribute to an ®B helix, now form a ¯-strand together with residues in the N-terminus and the activation loop. This leads to a misalignment of the catalytic residues and prevents ATP binding by steric hindrance of Thr60 in the nucleotide binding loop. By consequence, activation of MSK1 requires rearrangement of the nucleotide binding loop, the activation segment and the ®C helix. As to date, such a major conformational change has not been reported for MSK1. However, this type of conformational change has been confirmed for PKB, whose catalytic domain resembles MSK1 (340).
2.3.2.2 MSK expression and function
MSK1 displays a neuronal expression pattern, which reflects its physiological function. Removal of Mg2+ from the medium induces repetitive excitatory discharges and results in neuronal cell death. Since ERK and MSK1 become activated during these discharges, ERK and MSK1 activation contributes to cell death. Absence of MSK1 in hippocampal neurons protects against this excitatory injury and prevents neuronal cell death (157). Another role for MSK1 lies in cocaine-induced locomoter sensitisation and place preference as studies with MSK1-/- mice indicated (120,44). Recently, a study by Sindreu et al indicated that MSK1 also plays a role in fear conditioning via the Ca2+-AC-ERK-MSK1 pathway (334). MSK2 expression occurs predominantly in adipose tissue, brain, heart and placenta (279). But no reports have indicated a specific phenotypical function for this kinase.
Subcellularly, MSKs reside in the nucleus, where they can phosphorylate histone H3, HMG14 and transcription factors like CREB, NF·B and ER81 (398,342,340,88,11,12,136). ER81 (Ets-related protein) regulates ontogenesis and breast tumour formation. p38-dependent activation of MSK1 induces phosphorylation of ER81 and its co-activators (p300 and CBP) and thereby enhances transcription (174). MSK1 also enhanced the suppressive effects of As2O3 (a potent inducer of apoptosis in leukemic cells) on the growth of primary leukemic progenitors from CML patients. Finally, it also induced c-fos, junB, mkp1 and nurr1 transcription (186,238).
2.3.3 MAPK-interacting kinases (MNK)
The MAPK-interacting kinases (MNKs) consist of MNK1 and MNK2, the only isoforms in mouse. In humans, there exist four isoforms: alternative splicing for MNK1 generates MNK1a and MNK1b and for MNK2, MNK2a and MNK2b. MNK isoforms differ only in their last exon. Translation of the last exon of MNK1a results in 89aa, whereas translation of MNK1b gives rise to 12aa (223). Because of this reduced C-terminal end, MNK1b lacks the ERK docking site and hence remains unresponsive to mitogenic stimuli. The short C-terminal end also caused the loss of the NES signal in MNK1b, but since it retained the NLS signal, it resides mainly in the nucleus, in contrast to MNK1a. In the nucleus, it can phosphorylate the nuclear fraction of eIF-4E, which may affect the translation of mRNA, and/or transport mRNA out of the nucleus (270).
MNK2 shares 94% homology with murine MNK2 and 71% human MNK1. Due to differential splicing MNK2a receives 80 residues, while MNK2b possesses an additional 29 residues (338). Although both MNK2a and MNK2b lost their NES signal, only MNK2b resides in the nucleus. This means that additional mechanisms ensure cytoplasmic localisation for MNK2a (223). Furthermore, binding of ERK to MNK2 protects ERK from dephosphorylation and inactivation, whereas Mnk1 cannot contribute to ERK stabilisation (275).
In contrast to RSKs and MSKs, MNKs posses a single
kinase domain, which resembles the CTK domain of RSKs and MSKs, or
the kinase domain of Ca![]() /Calmodulin-dependent kinases. Despite the similarity to the latter,
MNKs do not become activated by Ca
/Calmodulin-dependent kinases. Despite the similarity to the latter,
MNKs do not become activated by Ca![]() /Calmodulin-inducing signals (223,307).
Instead, mitogenic and stress stimuli can elicit MNK activation: MNK1
mainly responds to stress, because it contains a LARRR MAPK binding
site which provides higher affinity towards p38 than towards ERK.
MNK2 responds better to ERK signalling because of its LAQRR motif
that renders higher affinity towards ERK. Neither of the MNKs can
bind JNK (275).
/Calmodulin-inducing signals (223,307).
Instead, mitogenic and stress stimuli can elicit MNK activation: MNK1
mainly responds to stress, because it contains a LARRR MAPK binding
site which provides higher affinity towards p38 than towards ERK.
MNK2 responds better to ERK signalling because of its LAQRR motif
that renders higher affinity towards ERK. Neither of the MNKs can
bind JNK (275).
Activation of MNKs requires a conformational change that displaces the Phe of the DFD motif within the ATP binding site, so that ATP can bind there. The DFD motif constitutes a unique feature of MNKs compared to other kinases, where the motif constitutes DFG residues (223,112). Another feature of MNKs is the Zinc binding motif (175).
2.3.3.1 Substrates
Both MNKs can phosphorylate eIF-4E on Ser209. This occurs after GF, hormone or mitogen stimulation and in case of MNK1 requires the scaffold protein eIF-4G. Phosphorylation of eIF4-E enhances cap-dependent translation. MNK1b displays a higher basal activity towards this substrate compared to MNK1a and suggests that different isoforms of the same kinase may regulate different levels of activity towards their substrates (223,264,175).
The three fusion proteins expressed in AML all stabilise MNK1 and thereby induce proliferation. All-trans retinoic acid (ATRA) also contributes to the decrease in half life of MNK1 and hence increases cap-dependent translation (401).
MNK1 can phosphorylate Sprouty2 (Spry2), a negative feedback modulator for RTKs (see section 1.2) during organogenesis in Drosophila and mammals. Sprouty proteins can become phosphorylated on their Tyr or Ser residues, but MNK1 only phosphorylates its Ser112 and Ser121 and thereby stabilises Spry2 upon GF stimulation (EGF, FGF). Prevention of phosphorylation or a mutation of phosphorylable Ser residues in Spry2 results in increased Tyr55 phosphorylation and subsequent degradation of Spry2 (80).
A yeast-two-hybrid screen identified MNK2 as a selective partner for binding to oestrogen receptor-¯ (ER-¯). MNK2 does not bind oestrogen receptor-® nor RSKs, which can phosphorylate ER-¯ . However whether ER-¯ is a substrate for MNK2 remains uncertain (338). Other substrates for MNKs include PLS2 and hnRNPA1 (223).
2.3.3.2 Function
In general, MNKs mainly influence protein synthesis. For example, angiotensin II-induced protein synthesis in vascular smooth muscle cells requires MNK1 (171). MNK1 may also play a role in the TNF-® production and stress responses (223). Furthermore, MNK1 is highly expressed in 25% of all acute myeloid leukemia cases (AML), where it affects myeloid differentiation. Because of the increase in MNK1, the amount of phosphorylated eIF-4E increases as well and causes deregulated proliferation and tumourigenic transformation. In contrast, inhibition of MNK1 enhances differentiation (401).
Ueda and coworkers investigated the role of MNK1 and MNK2, by constructing KO mouse models. Both single and double MNK KO mice were viable, fertile and displayed no apparent abnormalities. This contrasts with the diet-dependent effects observed in Drosophila, where MNK1 and MNK2 are required for cell growth and ontogenic development (381,120,295,275).
2.3.4 MAPKAPK 2 and 3
The remainder of the MAPKAPKs consists of MK2, MK3, MK4 and MK5. Although they all share the name MK, their expression pattern varies considerably among different organisms. All vertebrates express MK2, whereas only birds and mammals express MK3. Worms and fruit flies lack MK3 and sea urchin holds the exclusive right to synthesise MK4. Aside from the report in which MK4 was discovered, no other papers have since appeared to study its function (264,117). The eukaryotic yeast contains functional homologues of the mammalian MKs. These homologues also belong to the CaMK family and display most similarity to MKs in their catalytic domains. The yeast MKs consist of Rck1 and Rck2 in budding yeast and Mkp1 and Mkp2 in fission yeast. All these homologues become activated by Hog1 and Sty1, the p38 homologues in S. cerevisiae and S. pombe respectively (14).
This section mainly addresses MK2 and to a lesser extent MK3 because of fewer information available. Section 2.4 handles about MK5.
|
|
||
|
|
|
|
|
|
||
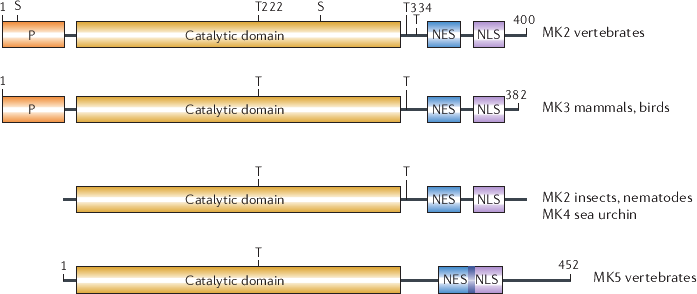
Figure: Overview of the domain structure of MAPKAPK2, 3, 4, 5. All kinases share a catalytic domain with a conserved phosphoacceptor site and a NES/NLS sequence. In addition, MK2 and MK3 also possess a Pro-rich N-terminal region for protein-protein interaction and regulatory phosphoacceptor sites in the hinge region between the N-terminal and C-terminal lobe. See text for details. The Figure is adapted from (117).
2.3.4.1 General
MAPKAPK2 or MK2 was isolated from skeletal muscle as
a kinase activated by the ERK pathway. In mice and humans, there
exist two isoforms of MK2 (46kDa and 54kDa) generated by differential
splicing and which vary in their C-terminal part (202,133).
Both of them bind p38![]() and become activated by it (117).
MK3 or 3pK, was discovered at the same time by two groups: once in a
yeast-two hybrid screen as a binding partner for p38 and
independently by sequencing a tumour suppressor region in human
chromosome 3 and isolating the associated kinase, 3pK. ERK, JNK and
p38 could all phosphorylate and activate the kinase (264).
MK2 and MK3 become mainly activated by stress stimuli. However,
mitogenic stimuli can also activate MK3. This activation occurs
faster than with stress stimuli and results in the presence of active
MK3 in the nucleus and the phosphorylation of substrates there (417).
Clifton and coworkers reported that MK2 when overexpressed in COS
cells was also susceptible to mitogenic stimuli, but that endogenous
MK2 exclusively responded to p38 (61).
Activated MK2 can phosphorylate targets that comprise the hydXRX(2)
S-motif (where ``hyd'' indicates any hydrophobic
residue) (330,262,201).
These targets include transcription factors (like E47, heat shock
proteins, CREB and ATF1), members of the Polycomb group (PcG),
lymphocyte-specific protein 1 (LSP1) and tyrosine hydroxylase. Some
of them are discussed in section 2.3.4 (202,262).
and become activated by it (117).
MK3 or 3pK, was discovered at the same time by two groups: once in a
yeast-two hybrid screen as a binding partner for p38 and
independently by sequencing a tumour suppressor region in human
chromosome 3 and isolating the associated kinase, 3pK. ERK, JNK and
p38 could all phosphorylate and activate the kinase (264).
MK2 and MK3 become mainly activated by stress stimuli. However,
mitogenic stimuli can also activate MK3. This activation occurs
faster than with stress stimuli and results in the presence of active
MK3 in the nucleus and the phosphorylation of substrates there (417).
Clifton and coworkers reported that MK2 when overexpressed in COS
cells was also susceptible to mitogenic stimuli, but that endogenous
MK2 exclusively responded to p38 (61).
Activated MK2 can phosphorylate targets that comprise the hydXRX(2)
S-motif (where ``hyd'' indicates any hydrophobic
residue) (330,262,201).
These targets include transcription factors (like E47, heat shock
proteins, CREB and ATF1), members of the Polycomb group (PcG),
lymphocyte-specific protein 1 (LSP1) and tyrosine hydroxylase. Some
of them are discussed in section 2.3.4 (202,262).
MK2 and MK3 are ubiquitously expressed with high levels in spleen, lung and skeletal muscle, but except for skeletal muscle, MK3 expression is tiny compared to MK2 (303). Both kinases resemble respectively p38® and p38¯ in their activity profiles and redundancy. MK3 can partially overcome the loss of MK2, as p38¯ can during loss of p38®. Similar to p38®, MK2 displays the major activity, whereas p38¯ and MK3 are dispensable for signalling (61,303).
2.3.4.2 Sequence and Structure
2.3.4.2.1 Sequence features
MK2 and MK3 share a higher similarity in primary sequence (75%), domain content and kinetics of nuclear export with each other than either of them with MK5 (~45%) (61,328,264). In the consensus sequence, they both require a bulky hydrophobic residue at ``n-5'' (where ``n'' means the phosphorylation site) and an Arg at ``n-3''. As for the phosphoacceptor site itself, MK3 can better tolerate the substitution of Ser at position ``n'' compared to MK2 (61,143). MK2 and MK3 possess an auto-inhibitory domain and NES/NLS sequences (resp.356-368/373-389) in their C-terminal domain, where the latter allows for p38 docking (143,117). Similar to p38 binding, removal of the C-terminal part of MK2, which includes the auto-inhibitory domain (res. 328-364), results in predominant cytoplasmic presence. Even more, it leads to the constitutive activation of MK2. The same holds for MK3 (417,143,261,359). The shortest splice form of MK2 lacks the NLS, with the binding site for p38, and a NES. This means that the 46kDa MK2 cannot bind p38 or respond to its signalling (202).
2.3.4.2.2 Structural features
|
|
||
|
|
|
|
|
|
||
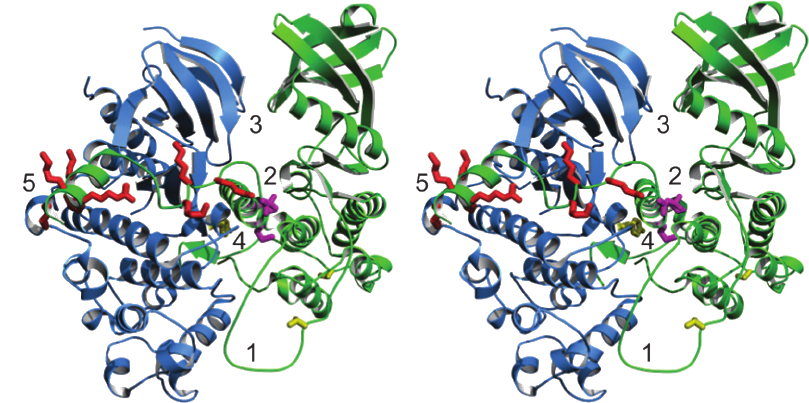
Figure 2.6: Structure of the interaction between p38® (blue) and MK2 (green). The five intermolecular interactions are identified by numbers 1 to 5. Additionally, the bipartite NLS signal for MK2 (red), the MK2 NES (magenta) and the regulatory phosphorylation sites on Tyr182 in p38® and Thr222 and Thr334 in MK2 (yellow). For more information on the structure of MK2 see section 2.3.4. Figure from (396).
There exist several crystal structures of MK2, one of the most recent has been discussed in section 2.1. Here, some extra features will be elucidated. In the inactive Apo MK2 structure, the long ®-helix of the auto-inhibitory domain blocks the the substrate binding site and reveals a wide open ATP pocket (240). When low affinity inhibitors (ADP, staurosporine) block MK2 in the active conformation, the ATP binding site adopts a closed conformation (382,7). On the other hand, when a high affinity inhibitor binds the ATP site, the auto-inhibitory domain is thought to flip aside and interact with nuclear export receptors via its NES sequence (143).
Recently, White and colleagues resolved the crystal structure of the heterodimer p38:MK2. Fi-gure 2.6 illustrates how the kinases bind each other head to head and how they present their catalytic sites on the approximate same side of the heterodimer. They form extensive intermolecular interactions that involve the p38® docking site as well (396). This provides us with an idea as to how kinases can interact with each other.
2.3.4.3 Functions for MK2 and MK3
2.3.4.3.1 Protein stabilisation
The interaction between p38 and the C-terminus of MK2 leads to the stabilisation of p38. Hence, depletion of MK2 results in decreased levels of p38 MAPKs in several tissues (202,303,133). On the other hand, lack of p38® results in decreased expression levels of MK2 as well, as MK2 expression can be reestablished upon reintroduction of p38® . This implies that both kinases exert a reciprocal stabilisation on each other (202,349).
2.3.4.3.2 Cellular localisation
In non-stressed cells, MK2 and MK3 reside in the nucleus, and both translocate to the cytoplasm upon activation by stress stimuli (105,201,24,130). Translocation of MK2 requires the phosphorylation of the C-terminal residue Thr317 by p38. Subsequent nuclear export depends on the Crm1 family of proteins and involves transport of p38 into the cytoplasm as well. Stress stimuli also induce nuclear export of ectopically expressed MK3, so that upon this stimulus, activated MK3 is only found in the cytoplasm (320,264,117).
2.3.4.3.3 Stress response
Stress stimuli can activate the p38 pathway which leads to the activation of MK2. Stimuli that can elicit such activity include amongst others LPS, Toll-like receptor ligands (TLRL) and heat shock. LPS stimulation affects the production of pro-inflammatory cytokines (see further). TLRL, produced by many pathogens, bind to Toll-like receptors (TLR) and lead to the activation of the NF·B, ERK and p38 pathways, which results in the production of TNF-®. This TLRL-induced p38 activation and TNF-® production depends on MK2 activity (369). Survival after sublethal heat shock involves MK2 activity since MK2 deficient cells displayed an impaired disaggregation of endogenous Hsp25, a delayed kinetics for insolubilisation of Hsp25, and increased apoptosis. Recovery after lethal heat shock was not affected by MK2 since MK2 KO cells produced a fast and complete insolubility of Hsp25 (385). Responses to stress (like sublethal heat shock or H2O3) disrupt the actin microfilament network in the cell and induce the activation of MK2. Subsequent phosphorylation of Hsp25/27 by MK2 stimulates the polymerisation of actin and thereby helps to stabilise the microfilament network (61,385). MK3 can phosphorylate Hsp25 as well, but near complete loss of Hsp25 phosphorylation in MK2-deficient cells suggests that MK3 is not essential for Hsp25 phosphorylation (133).
MK2 can also phosphorylate 14-3-3³ on Ser58. The phosphorylation of this residue is assumed to impair dimerisation with Raf1, as simulated by the behaviour of an S58D mutant. The precise role of this interaction remains unclear, but since 14-3-3³ can interact with leukocyte-specific adhesion molecules which play a role in neutrophil adhesion, this interaction may indicate a role during inflammation (284).
2.3.4.3.4 Cell migration
Cells deficient in MK2 display defects in filopodium and microspike formation as well as in cell migration. Rescue of cell migration can be elicited by introduction of MK2 that contains an intact N-terminal Pro-rich region and that is catalytically active. The role of MK2 in cell migration may be explained by its interaction with Hsp25/27. The non-phosphorylated form of Hsp25/27 inhibits actin polymerisation at the barbed end, whereas phosphorylation by MK2 blocks this inhibition (202).
In endothelial cells, MK2 seems the missing link between p38 and LIMK1 that regulate cytoskeletal rearrangements during VEGF-A induced angiogenesis. Although p38 can phosphorylate LIMK1, it does not seem to activate it. On the other hand, MK2 can phosphorylate and activate LIMK1 which leads to the formation of stress fibers. This process involves the inhibition of the polymerising activity of cofilin (198).
2.3.4.3.5 Cell cycle checkpoints and cancer
When DNA damage is detected in a cell, Chk1 and Chk2 become activated. They phosphorylate cdc25 phosphatases and thereby promote their binding to 14-3-3¾. This inactivation of cdc25 prevents entry into the M-phase. A similar inactivation of cdc25 can be elicited by MK2 activation in response to UV irradiation, where MK2 can phosphorylate cdc25 on S169, S323, S353 and S375. Because of this functional similarity to Chk1 and Chk2, MK2 is also called Chk3 (233,214).
In normal cells, DNA damage activates p53, which subsequently activates a checkpoint for DNA integrity or induces apoptosis when the cell cycle has progressed too far. In cells devoid of p53, DNA damage may trigger the activation of Chk1 and p38-induced activation of MK2. Inhibition of these kinases by UCN-01 prevents cell survival and leads to regression of mouse tumours (296). This is consistent with reports of increased MK2 activity in breast cancer.
Another function of MK2 lies in the phosphorylation of tuberin (TSC2), a tumour suppressor protein. Hamartin (TSC1) and tuberin (TSC2) both negatively regulate cell growth by inhibiting protein synthesis. Deletion of either protein results in embryonic lethality, and mutation of either protein accounts for 50% of the cases with Tuberus Sclerosis complex (TSC). This autosomal dominant genetic disease is characterised by the formation of benign hamartomas in many organs resulting in mental retardation, seizures and autism. Phosphorylation of TSC2 by MK2, but not MK5, increases binding of TSC2 to 14-3-3 proteins in response to serum and anisomycin. This binding inhibits the function of TSC2 (218).
2.3.4.3.6 Transcriptional and post-transcriptional regulation
MK2 and MK3 can bind and phosphorylate the basic helix-loop-helix transcription factor E47. E47 plays a role in differentiation and lineage commitment in B-cells. Its phosphorylation represses E47-induced transcriptional activity (260). MK2 can also phosphorylate transcription factors ER81, SRF and CREB (117).
MK3 can affect chromatin remodelling by phosphorylating members of the Polycomb Group (PcG), which repress transcription complexes during proliferation and differentiation. Overexpression of MK3 leads to increased phosphorylation of Bmi and other members of the PcG and results in their dissociation from chromatin. This may relieve the transcriptional repression of e.g. p14ARF in the Cdkn2a/INK4A locus (390).
Psoriasis skin also expresses increased MK2 activity and thereby displays increased production of TNF-® (182). Similarly increased pro-inflammatory cytokine production upon LPS stimulation is impaired in MK2 deficient cells (202,133).
MK2 can regulate the stability or translation efficiency of mRNA transcripts that contain class II AU-rich elements (ARE). It does this by phosphorylating proteins that bind the ARE sequences. For example, in LPS-treated macrophages, MK2 phosphorylates hnRNP40A0 at Ser84 and thereby destabilises MIP2, COX-2 and TNF-® mRNAs. Treatment with the p38 inhibitor SB203580 prevents this destabilisation. Apparently, MK3 and MK5 can phosphorylate hnRNPA0 in vitro, which may explain the weak phosphorylation of hnRNPs in LPS-treated MK2 KO macrophages (305). Another example of an ARE binding protein regulated by MK2 is TTP (also called Nup75, TIS11, G0S24, ZFP-36). Phosphorylation by MK2 on Ser52 and Ser178 inactivates TTP and increases its stability (59). Cells that lack MK2 express less TTP and therefore contain increased amounts of pro-inflammatory cytokine mRNA molecules (Cox2, IL-6, IL-8, TNF-®). This explains why TTP KO and TTP/MK2 double knockout (DKO) mice display autoimmune diseases (202,133,145). Although MK3 can partially stabilise TTP in absence of MK2, it cannot compensate for the loss of MK2 completely (303).
2.3.4.3.7 Mouse models
As to date, several MK2 KO mouse models have been constructed. MK2 deficiency yields fertile, viable, normally sized mice which display no specific behavioural defects, nor chromosomal alignment defects or spindle abnormalities during mitosis. However, MK2-deficient mice demonstrated an increased sensitivity to high-salt feeding and resistance to LPS-induced shock. Additionally, they produced reduced amounts of cytokines in spleen cells and serum (201,117). Because of impaired cytokine synthesis, MK2 KO mice became susceptible to inflammatory diseases like collagen induced arthritis (CIA), pancreatitis and Lysteria infection (372,213,138). However, these mice had a better outcome after heart and brain ischemia compared to littermate controls (120,330,395).
Recently, a report on the construction of MK3 KO mice was published. These mice exhibit no particular phenotype and display no significant changes in cytokine production, nor decreases in p38 or TTP levels. The DKOs for MK2 and MK3 show no defect in embryonic development, which was unexpected because both MK2 and MK3 interact with the polycomb complex. The mice are viable, fertile and behave normally. However, macrophages from these mice display an even further reduced TNF synthesis, compared to MK2 alone (303). This contrasts to the DKO of MK2 and MK5, which displayed no specific phenotype, but showed no additional reduction in TNF-® synthesis either (328). These results indicate that for the treatment of inflammatory diseases, it would be favourable to use inhibitors that target both MK2 and MK3.
The phenotype from DKO mice lacking MK2 and TTP resembled the phenotype of the KO of TTP alone. The mice displayed cachexia, a ``Kangaroo'' hunched posture, reddened and swollen joints and moderate to severe infiltration of neutrophils and plasma cells in several tissues, due to the stabilisation of many pro-inflammatory cytokine mRNAs (145).
2.4 MAPK-activated protein kinase 5 (MK5)
The last of the MAPKAPKs, MAPKAPK5 (MK5) or its human analog p38-regulated and activated protein kinase (PRAK) was discovered simultaneously by two groups, approximately 10 years ago. New and coworkers searched a human placental cDNA library that encoded the MAPKAPK sequence LXTPQFTPYYVAP and found a protein of 54kDa comprising 471 residues. The kinase could be activated by p38, so they called it PRAK (262). Almost simultaneously, Ni and colleagues discovered MK5 by searching the EST database with the nucleotide sequence of MK2 as query sequence. The fragment they found was used as a probe in a murine spleen cDNA library and led to the identification of a protein of 54kDa with 473 residues (264). The discrepancy in the amount of residues reported by both groups is due to differential splicing. Mice express 5 isoforms and humans express 2 isoforms of MK5. These two isoforms only differ in two residues in their C-terminal end, and it remains unknown if these isoforms contribute differently to a cellular function (117). Besides murine and human species, MK5 is expressed in all vertebrates except for C. elegans and Drosophila spp. Within mammalia, MK5 appears in most tissues including the heart, brain, lungs, liver, skeletal muscle, kidney, testis and pancreas (123,328,264).
2.4.1 Sequence
MK5 resembles other MAPKAPKs by approximately 20-30%, while it shares approximately 45%, 46% and 44% aa identity with MK2, MK3 and MK4 respectively (35,264). This contrasts to the high homology between MK2 and MK3, and is probably due to the unique C-terminal tail of MK5.
The N-terminal region of MK5 lacks the Pro-rich protein-protein interaction domain, present in MK2 and MK3 (see Fig 2.5). In contrast, the catalytic domain resembles the one in MK2 and MK3 (in sequence and structure), comprising a conserved Thr182 in the LMTP motif of the activation loop. This residue becomes phosphorylated by p38 and results in increased kinase activity. In contrast to MK2 and MK3, MK5 lacks additional regulatory phospho-sites in the hinge region. In MK2 and MK3, these regulate the interaction of the auto-inhibitory domain with the catalytic site. Although MK5 contains a putative auto-inhibitory ®-helix, its possible role in MK5 remains unknown (262).
Similar to MK2 and MK3, MK5 also carries NES and NLS sequences. In MK5, these sequences are fused together but still allow shuttling between the nucleus and the cytoplasm. The NLS sequence of MK5 overlaps with the p38 binding site and consequently p38 binding prevents nuclear localisation (see section 2.4.2). Another regulatory sequence in MK5 is the partially conserved PKI-like NES consensus sequence, which binds the inhibitor of PKA (PKI) and exports bound protein out of the nucleus. Mutation of one of the four spaced hydrophobic residues that lie within this consensus sequence, Leu337, destroyed the functional NES but surprisingly also resulted in a constitutive active protein, indicating its importance in structural rearrangements for MK5 activity (324).
Finally, the unique C-terminal tail of MK5 shows no resemblance to any of the other MKs. This tail is most likely involved in substrate binding (see 2.4.2), but how it is folded and what its impact is on MK5 its function remains difficult to predict. Since the 3D structure of MK5 remains unresolved and in silico modelling of the protein is difficult due to its unique C-terminal tail.
2.4.2 Interaction partners
|
|
||
|
|
|
|
|
|
||
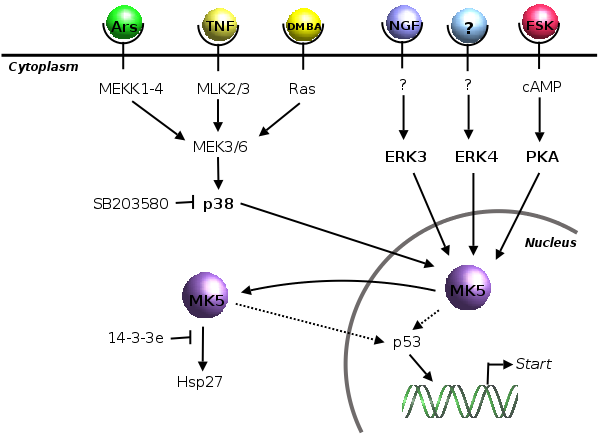
Figure 2.7: Overview of the known in vivo interaction partners for MK5 discussed in section 2.4.2.
Figure 2.7 illustrates the known in vivo interaction partners of MK5, which will be discussed in this section.
2.4.2.1 p38
Along with MK5 its discovery, New et al. reported the first in vivo interaction partner for MK5, namely p38. Although they did not prove the in vivo interaction between the two proteins, they studied which stimuli could induce the p38-dependent PRAK activation. Stimuli that strongly activated PRAK included mostly stress stimuli like arsenite, anisomycin, H2O3 and TNF-®, whereas unlike MK3, stimulation with IL-6 and the family of growth factors (EGF, FGF, PDGF) did not affect the activity of PRAK.
The interaction between p38 and MK5 resulted in a redistribution of MK5 within the cell. One report found MK5 predominantly in the cytoplasm in unstimulated cells (261), the other found MK5 mainly in the nucleus (324). Both reports used endogenous MK5 as well as ectopically expressed MK5 and agreed that MK5 shuttled between the nucleus and the cytoplasm in unstimulated cells. They also agreed on the cytoplasmic relocalisation of MK5 upon stress stimuli and the necessity for Thr182 phosphorylation to induce such redistribution. Finally, both groups used HeLa and HEK293 cells for their studies. The reports differ in the type of MK5 antibody they used and the sequence of the NES reported (324,261). The antibody that New and coworkers used was later shown to crossreact with MK2, which may explain why they found a different subcellular location for MK5 in unstimulated cells (261).
|
|
|||
|
|
|
|
|
|
|
|||
 A
A B.
B. Figure 2.8: Two possible models for the interaction between p38 and MK5. Model A depicts the majortiy of PRAK molecules in the cytoplasm by docking to cytoplasmic p38¯. The p38 site overlaps with NLS therefore PRAK interaction masks this site and thereby prevents nuclear import of PRAK (261). In model B, p38 can increase nuclear export of MK5 via 2 different mechanisms: exposing the NES which requires phosphorylation of Thr182 and by masking the NLS by binding to MK5 and interfering with the function of the NLS (324).
Nevertheless, the interaction between MK5 and p38
remains a controversial issue, since yet another report showed that
the absence of MK5 did not affect the availability of p38, whereas
MK2 deficiency led to reduced amounts of p38 in heart, lung and
muscle cells. Even more, this report showed that p38 did not increase
the activity of MK5 upon stimulation, whereas p38 could activate MK2
under similar circumstances (328). A
later report by Seternes and coworkers stated that they were unable
to pulldown MK5 and endogenous p38![]() together, jeopardising a possible p38
together, jeopardising a possible p38![]() -MK5 interaction in vivo (325).
However, a recent report by Sun et al proved that p38 can activate
PRAK in vivo and that this leads to increased PRAK activity (see
2.4.2) (351).
This indicates that cellular specificity and the presence or absence
of particular stimuli determine whether or not these two kinases
interact.
-MK5 interaction in vivo (325).
However, a recent report by Sun et al proved that p38 can activate
PRAK in vivo and that this leads to increased PRAK activity (see
2.4.2) (351).
This indicates that cellular specificity and the presence or absence
of particular stimuli determine whether or not these two kinases
interact.
2.4.2.2 ERK3
Two reports described a p38-independent but specific interaction between ERK3 and MK5 (320,325). Both reports agree that ERK3 specifically interacts with MK5, and that it has no affinity for MK2 or MK3. The two kinases interact via residues 330-340 on ERK3 and residues 423-473 on MK5. Furthermore, interaction results in nuclear export of both proteins and does not require a phosphorylated Thr182 on MK5, nor the activity of either kinase to elicit this relocation. Although both groups used HeLa cells and ectopically expressed kinases, they disagree on the mechanisms of this nuclear export. Seternes et al report Crm1-dependent relocation, whereas Schumacher and colleagues report the contrary. In addition, there exists also some discrepancy in the mechanism of activation. One report shows that ERK3 can phosphorylate and activate MK5 and that ERK3 is a substrate for MK5 (325). The other report shows that catalytic activity of ERK3 is not required for MK5 translocation and activity, but that catalytic activity and phosphorylation of Thr182 is necessary for activation of MK5 (320). Finally, similar to the p38-MK2 interaction, ERK3 can stabilise MK5 and vice versa, as knockdown of one kinase elicits a reduced expression of the other (325). The role of this interaction may lie in cell proliferation since ERK3 expression in human carcinoma cell lines displayed decreased cell proliferation, in accordance with an earlier report that showed inhibition of cancer cell growth, migration and invasion upon overexpression of ERK3 (71,325).
2.4.2.3 ERK4
The interaction between MK5 and ERK4 was proven by the two groups that reported the ERK3-MK5 in vivo interaction (426,187). Similar to ERK3, residues 326-340 of ERK4 bind the C-terminal 423-472 residues of MK5 and the interaction leads to Crm1-dependent nuclear export of both proteins. In contrast to MK5, Kant et al reported mainly cytoplasmic presence for ERK4, whereas Åberg et al reported exclusively cytoplasmic presence. Although they acknowledged active temperature-dependent import of ERK4 into the nucleus as well (426,187).
Since ERK4 can form heterodimers with ERK3, it may be possible that the ERK3 activity towards MK5 mentioned in a previous report by Seternes et al, occurs because of ERK3s interaction with ERK4 (426,187,325). This confirms the catalytic activity of ERK4 towards MK5. The importance of ERK3/ERK4 activity for MK5 activation is illustrated by the fact that knockdown of ERK3 and ERK4 resulted in an approximate 80% inhibition of MK5 activity (426). Also similar to ERK3, MK5 can phosphorylate ERK4 (187). Finally, the physiological importance of this interaction remains enigmatic as well, but it may play a role during development (426,187).
2.4.2.4 p53
Sun and coworkers described the first physiological function for MK5 as a tumour suppressor by activating p53 in a Ras-dependent way. Although there exist several ways by which Ras can induce senescence, the authors showed that here Ras-dependent activation of p38 was required. This in turn activated PRAK, which then phosphorylated p53 on Ser37, thereby stimulating its activity (351). The activation of PRAK by senescence was measured by its activity towards Hsp27 in BJ cells, when these cells were exposed to Ha-RasV12 or caMKK3/MKK6. Under such circumstances, PRAK activity was prevented by treatment with SB203580. Similarly, PRAK knockdown by shRNA prevented G1 arrest upon UV irradiation or it induced immediate transformation of the cells upon introduction with Ha-RasV12. This may indicate that PRAK knockdown can functionally replace the negative regulator of p53, MDM2, in Ras-induced senescent cells. On the other hand, overexpression of PRAK alone led to decreased proliferation rates, but did not suffice to induce premature senescence on its own. This indicates that PRAK needs additional upstream signals to exert its tumour suppressing effect during cellular senescence (351). This group also found that MK5 deficient mice were more susceptible to chemical-induced skin cancer compared to wild-type mice (see 2.4.3).
2.4.2.5 14-3-3²
2.4.2.6 Heat Shock protein 25/27
14-3-3 proteins regulate several processes in the cell, but play a prominent role during the cell cycle, cell survival and apoptosis. They exist as seven isoforms which dimerise and recognise specific phosphorylated residues on their substrates (245,126,141,283). Recently, Tak et al reported an in vivo interaction between MK5 and 14-3-3² upon stimulation of HeLa cells with TNF-®. Binding of 14-3-3² required residues 1-308 of MK5, and reports the first partner that binds to the N-terminal region. Binding of 14-3-3² is independent of the phosphorylation of Thr182, but inhibits MK5 its activity towards Hsp27. This in turn negatively regulates the polymerisation of G-actin into F-actin and inhibited cell migration (355).
Hsp25/27 functions as a chaperone for protein folding, but in addition it binds to the barbed end on the F-actin filament to prevent further polymerisation. From its discovery, MK5 has been reported to phosphorylate Hsp25/27 at Ser15, Ser78 and Ser82, but there existed considerable doubt whether the interaction occurred in vivo as well (320,261,328,325). Recently, Tak et al demonstrated by co-immunoprecipitation that MK5 could interact with Hsp27 and phosphorylate it in vivo [355, S. Kostenko, unpublished results].
2.4.2.7 In vitro interaction partners
2.4.2.7.1 ERK2
Similar to p38®/¯ but in contrast to JNK, ERK2 could phosphorylate and activate MK5 in vitro. This contradicts reports that show that MK5 does not respond to GF stimuli, which signal via ERK1/2 (328,264). As to date, no in vivo studies have confirmed such an interaction.
2.4.2.7.2 Cytosolic phospholipase A2 (cPLA2)
cPLA2 hydrolyses phospholipids and thereby synthesises arachidonic acid (AA) and platelet activating factor (PAF). These products serve as precursors for leukotrienes, prostaglandines, thromboxanes and prostaglandines. cPLA2 resides in the cytosol as well as in the perinuclear region, the endoplasmic reticulum and the Golgi apparatus (144,54). Hefner and coworkers demonstrated that MNK1 and MK5 could phosphorylate cPLA2 on Ser727 in platelets and that treatment with p38®/¯ inhibitor could prevent such phosphorylation. At present, a putative in vivo interaction remains unresolved, as no co-immunoprecipitation with MK5 and cPLA2 has been performed (137).
2.4.2.7.3 Tyrosine Hydroxylase (TH)
Tyrosine hydroxylase catalyses the rate-limiting reaction in the biosynthesis of catecholamines (adrenalin, noradrenalin and dopamine), namely the conversion of L-tyrosine into L-DOPA. KO mice in TH display embryonic lethality, which illustrates the importance of TH in several physiological processes that are also regulated by dopamine e.g. food intake, addiction and control of movement (148). Several reports have indicated that MK5 can phosphorylate TH at Ser19 in vitro. Since the p38 inhibitor SB203580 prevented phosphorylation of TH at this residue in arsenite-treated cells, this suggests that PRAK may mediate the in vivo phosphorylation of TH. However, there exists no explicit proof for an such an interaction (375,374).
2.4.2.7.4 Myosin heavy chain
New et al reported that MK5 can phosphorylate the heavy chain of myosin in vitro (262). Later New et al refer to their first report, as MK5 that phosphorylates the regulatory light chain of myosin II in vitro (261). This may be in agreement with the discovery of MK4 as a myosin II regulatory light chain kinase (200). Furthermore, a blast search of the peptide used by Ni et al reveals that the sequence resembles myosin regulatory light chain. So far, no other reports have confirmed this interaction in vivo.
2.4.3 MK5 transgenic mice
Since its discovery, approximately 10 years ago, the biological role and significance of MK5 still remain enigmatic. Several attempts to shed light on its function revealed only very little information. This section describes the in vivo attempts to pinpoint possible functions for MK5 by means of transgenic mice. To date, there exist three different approaches towards mouse models for MK5 :
-
In two KO models exon 6 (aa131-161) was deleted. This exon codes for parts of the catalytic core of the kinase (subdomains VIa and VIb). The preliminary model was bred in a 129xC57BL/6 mixed background, whereas the other model was bred in a full C57BL/6 background (320,328).
-
The KO mouse model that lacked exon 6 was bred onto a MK2-deficient mouse line to yield a MK2/MK5 DKO mouse model (328).
-
In the last KO model, researchers deleted exon 8 and bred the mice in a 129xC57BL/6 mixed background (351).
Deletion of exon 6 in a mixed background resulted in
an inactive and unstable protein. The resulting mice seemed healthy
and fertile and revealed no behavioural or morphological
abnormalities. Since MK2 KO compromised immune responses, the spleen
cells of MK5 KO mice were stimulated with LPS to assess cytokine
responses (TNF-® , IL-6 or IFN-°)
during lack of MK5. Compared to MK2 deficiency, KO of MK5 produced no
alteration in cytokine responses, although a slight increase occurred
in IFN-° and IL12p40 levels, but not
in IL-6 levels (262,328).
Both MK5 and MK2 KO mice displayed a slight decrease in IL-10. The
protein levels of the upstream activator of MK5, p38, seemed
unaffected as well, arguing against a chaperon-like function for MK5
during p38 interaction, like the one demonstrated for MK2. The
MK2/MK5 double KO mice, displayed a similar reduction in TNF-![]() levels compared to MK2 KO mice alone. This suggests that MK5 does not
play an essential role in TNF-®
responses but it does not exclude the involvement of e.g. MK3 (328).
levels compared to MK2 KO mice alone. This suggests that MK5 does not
play an essential role in TNF-®
responses but it does not exclude the involvement of e.g. MK3 (328).
The same mutation bred to a full C57BL/6 background resulted in embryonic lethality with incomplete penetrance at E11. At this day during organogenesis, tissues where epithelial cells perform a secretory function, heart and skeletal muscle cells express maximum ERK3 levels. Of the embryos that survived past E12, only 50% of the expected embryos was homozygous for the MK5-/- mutation. The homozygous pups that developed to term were smaller in size, but developed normally and displayed no other abnormalities in morphology or histology up to 24 weeks. This suggests that MK5 in concert with ERK3 plays an essential role during development (320). Especially, in terms of incomplete penetrance the lack of MK5 might be interesting in terms of MK5 and cell migration.
Finally, deletion of exon 8 in a mixed background produced viable and fertile mice without health problems. However, upon skin treatment with dimethylbenzanthracene (DMBA), 7 of the 13 MK5-/- mice (53.8%) developed skin pappillomas, compared to 12 out of 30 MK5+/- mice and one in 12 of the WT mice (8.3%). In contrast, untreated mice remained tumour free for 2 years. Since heterozygous mice also displayed increased skin pappilloma formation compared to WT mice, tumour suppression by PRAK requires two copies of the gene to suppress tumour formation (haploinsufficient). In addition, PRAK expression levels between tumours and surrounding tissue were equal. However, in contrast to WT mice where senescence prevents pappilloma formation, the absence of senescence markers in the tumours of homo- and heterozygous MK5 deficient mice indicates that PRAK deficient tumour-induction occurs via suppression of senescence. Especially because senescence markers were present in the rest of the tissues of the transgenic mice (351).
3. The PKA signalling pathway
3.1 Introduction
In the late seventies, Fisher and coworkers reported that phosphorylase b kinase, the first protein kinase discovered, became phosphorylated by another kinase (331). This kinase received the name Protein Kinase A (PKA), or cAMP-Activated Protein Kinase and belongs to the AGC family of kinases (see 1.1.1). Like most other protein kinases, PKA possesses a regulatory domain and a catalytic domain. However, unlike most kinases, these two domains exist as separate polypeptides (263,216,140). This has two consequences: first, different types of subunits can form different PKA isoenzymes. Second, separate subunits allow for differential regulation of kinase activity by association and dissociation from each other. This section summarises how PKA works and what part it plays in the PKA signalling pathway.
3.2 Protein kinase A
The inactive form of PKA exists as a tetramer that consists of two regulatory (PKAR ) and two catalytic (PKAC ) subunits. Originally, inactive PKA heterotetramers were identified during ion exchange chromatography. Separated on the content of their regulatory subunits, they were described as type I subunits, which have a higher affinity for cAMP and reside predominantly in the cytoplasm, or as type II subunits that mainly associate with subcellular structures. The different cellular location of these two types implies their involvement in different cellular processes. Indeed, RI subunits control mainly cell proliferation, neoplastic transformation and G1/S phase progression, whereas RII subunits regulate differentiation, growth arrest and induction of apoptosis (300,41,74).
Later, molecular cloning revealed the existence of four isoforms for PKAR and four isoforms for PKAC. PKAR can exist as a homo- or heterodimer of RI® , RI¯ , RII® and RII¯ isoforms, where dimerisation occurs via disulphide bonds between the N-terminal dimerisation or docking domains (361,185,323). In addition to the docking regions, PKAR isoforms also possess a flexible linker, in which an auto-inhibitory domain lies, and two cAMP-binding domains (CBD), that contain a phosphate binding cassette (79). cAMP binds first to the -only- exposed CBD, the B-CBD, which then facilitates binding to the second CBD, the A-CBD. Binding of the four molecules of cAMP to PKAR results in a conformational change of the R-subunits and in the dissociation and activation of the C-subunits (185,361,363,323,191). The difference in conformation of the regulatory subunit of PKA when bound to cAMP or to the catalytic subunits is shown in Figure 3.1.
|
|
||
|
|
|
|
|
|
||
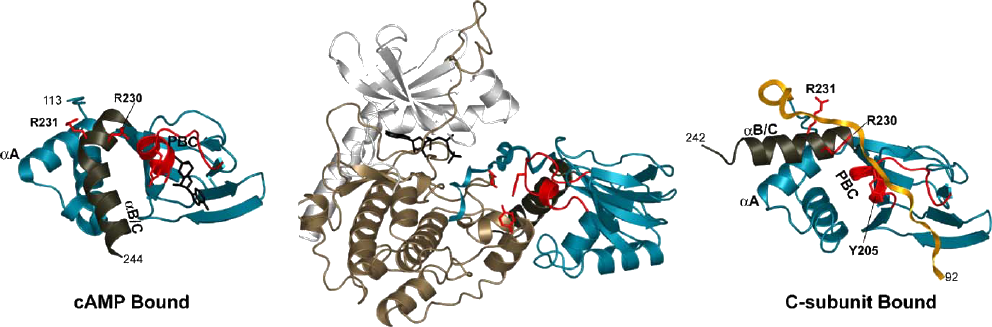
Figure 3.1: Conformational differences of the PKAI® (res.92-244) upon binding of cAMP (left) or the catalytic subunit (middle). The right panel shows the conformation of the regulatory subunit alone, when it is bound to the catalytic subunit (363).
There exist four isoforms for PKAC (40kDa): C®, C¯, C° and PrKX (185,323), where each catalytic isoform can bind any of the four regulatory isoforms within their hinge regions (185,332). The catalytic subunits consist of two lobes. The small lobe comprises residues 40-119 where ATP binds. The large lobe (res.128-300) mainly consists of ®-helices and serves as docking surface for peptide binding and facilitation of the phosphoryl transfer (364). After cAMP-induced activation and dissociation from the R subunits, the C subunits can translocate to the nucleus to phosphorylate a variety of nuclear substrates (216).
The human X chromosome-encoded protein kinase X,
PrKX, displays high expression levels in foetal tissues and adult
brain, kidney and lung. Lower expression levels can be found in the
placenta, heart, liver, skeletal muscle, pancreas and foetal liver.
PrKX differs from the other catalytic subunits of PKA in its
N-terminus, its preferential binding to the RI subunit and its
phosphorylation of RII![]() . There exists also a homologous gene on the Y chromosome, and
abnormal interchanges between the X and Y chromosome genes leads to
XX males and XY females (337,425,196).
. There exists also a homologous gene on the Y chromosome, and
abnormal interchanges between the X and Y chromosome genes leads to
XX males and XY females (337,425,196).
Because there exist different isoforms of PKAR and PKAC, each isoform contributes to specific pathways under specific circumstances. Expression of PKAR® and PKAC® occurs ubiquitously, whereas the ¯ subunits display a more restricted expression pattern. In general, neurons express high levels of RI¯, whereas especially striatal neurons, adipose and endocrine tissues express RII ¯. The three splice forms of PKAC¯ (C¯1, C¯2 and C¯3) are differentially expressed. C¯1 is ubiquitously expressed in all tissues at low levels, but is enriched in the hippocampus and the cortex. C¯2 can be found in the cortex and in the limbic structures of the brain, whereas C¯3 maintains a low expression level throughout the entire brain. The testis mainly express PKAC® (361,45).
The human X chromosome-encoded protein kinase X, PrKX,
displays high expression levels in foetal tissues and adult brain, kidney and lung. Lower expression levels can be found in the placenta, heart, liver, skeletal muscle, pancreas and foetal liver. PrKX differs from the other catalytic subunits of PKA in its N-terminus, its preferential binding to the RI subunit and its phosphorylation of RII®. There exists also a homologous gene on the Y chromosome, and abnormal interchanges between the X and Y chromosome genes leads to XX males and XY females (196, 337, 425).
3.3 The PKA pathway
Several signals (e.g. hormones and neurotransmitters, NTM) can activate the PKA pathway. For example when adrenaline binds to the adrenergic receptor, a GPCR, it induces a conformational change that results in the dissociation of a heterotrimeric G-protein linker from the receptor. The heterotrimeric G-protein dissociates into the G®-GTP monomer and the G¯°-dimer. The G®-GTP can stimulate (G®s ) or inhibit (G®i ) adenylyl cyclase (AC), the enzyme that converts ATP into cAMP. Four molecules of cAMP then bind to the two regulatory subunits of PKA and lead to the previously described activation and dissociation of the regulatory and catalytic subunits of PKA. The activated catalytic subunits can then translocate to the nucleus where they can phosphorylate a variety of substrates. The best studied nuclear substrate for PKA is the transcription factor Cyclic AMP Response Element Binding protein or CREB. CREB binds to the CRE element in the promoter region of cAMP-responsive genes, where phosphorylation of its Ser133 by PKAc greatly enhances transcription (216,178).
Along the PKA pathway, there exist several possibilities for regulation. For example, phosphodiesterases (PDE) can degrade the vice versa bonds of cAMP, thereby inhibiting the PKA activation (216,140). Furthermore, the catalytic subunit of PKA can bind to PKI, a protein kinase inhibitor with a strong nuclear export signal, that exports the catalytic subunit straight out of the nucleus. This prevents further cAMP-induced alterations in gene expression.
Another means for regulation of PKA activity consists of binding of the N-terminal docking region of PKAR subunits to the A Kinase Anchorage Protein, AKAP for short. AKAPs function like scaffold proteins (previously described in the MAPK pathway). They enhance the efficiency and specificity of interactions that take place within the PKA signalling pathway by gathering PKA, specific substrates or regulatory proteins, like PDEs, together in a specific place within the cell (361,151,323). Although, RI PKA has been described to bind AKAPs as well, mainly PKARII subunits seem to associate with AKAPs. This explains their predominant localisation to subcellular structures, whereas PKARI subunits remain soluble in the cytoplasm (361,300,151).
3.4 Physiological significance
Because of its cellular abundance, many reports described the importance of the PKA signalling pathway. Some examples:
-
An inactivating mutation in the PRKAR1A gene which encodes RI® leads to Carney complex, which is best modeled by RI®+/- mice (255,41). Furthermore, RI® anti-sense oligonucleotides (ODN) can inhibit colony formation and cell proliferation in a dose dependent manner in human breast cancer cells (344). Similarly, RI® ODN administration to DMBA-treated mice could delay the onset of tumour formation, reduce the number of tumours that developed and restore the previous levels of RII¯ (259).
-
Mice that lack the C¯1 subunit of PKA, display deficiencies in the late phase LTP response to high frequency stimuli due to impaired maintenance of gene transcription during LTP (207,286).
-
RII¯-/- mice display the inability to balance, an increased sensitisation to amphetamine (a drug that increases locomotion), an increased resistance to haloperidol/raclopride induced catalepsy and a lack of c-fos transcription after amphetamine administration (45,46).
-
A null mutation in the C
 subunit caused early
postnatal lethality in mice in a 88:12 C57BL/6:129SV/J background.
However, KO mice in a 50:50 C57BL/6:129SV/J background, survived more
frequently. In addition, the C KO
mice that survived weighed less due to overall decreased weight, which
was independent of deficiencies in organogenesis (336).
subunit caused early
postnatal lethality in mice in a 88:12 C57BL/6:129SV/J background.
However, KO mice in a 50:50 C57BL/6:129SV/J background, survived more
frequently. In addition, the C KO
mice that survived weighed less due to overall decreased weight, which
was independent of deficiencies in organogenesis (336). -
RI® disruption in mice results in a defective production and migration of the mesoderm and results in an increased basal activity of PKA in the KO mice. Crossing between RI® KO and C® KO mice, which carry the PKA R subunit, rescued these defects (155).
-
Mice that lack the sperm-specific catalytic subunit of PKA, C®2 or Cs are infertile due to a defective forward motility (268,336).
-
T-cells from Systemic Lupus Erythomatosis (SLE) patients display decreased IL-2 production, a cytokine for maintenance and propagation of T-cell function. These decreased levels may result from reduced PKA, and consequently, CREB activity. In addition, dephosphorylation of CREB by PP2A is increased in these cells (365).
3.5 Crosstalk between MAPK and PKA pathways
A cell integrates the many signals that arrive at each moment during its existence. This implies it should orchestrate the cooperation and interaction between different signalling pathways (crosstalk). This section summarises some of the crosstalk between the previously discussed MAPK pathway and the PKA pathway.
However, whether a particular pathway interacts with another pathway or not, depends on the cell type and the cellular context. To illustrate this, a report by Pearson and Cobb showed that increasing cAMP concentrations in proliferating NIH3T3 and C2C12 cells activated ERK5, whereas 24h serum deprivation of confluent cells inhibited ¯-FGF-induced activation of ERK1, ERK2 and ERK5 (276). Similarly, the expression of different isoforms of Raf resulted in different responses to crosstalk from the same PKA pathway. cAMP-induced PKA activity inhibited c-Raf and thereby the MAPK pathway, whereas the same activity promoted MAPK activation when B-Raf was expressed (151,278).
|
|
||
|
|
|
|
|
|
||
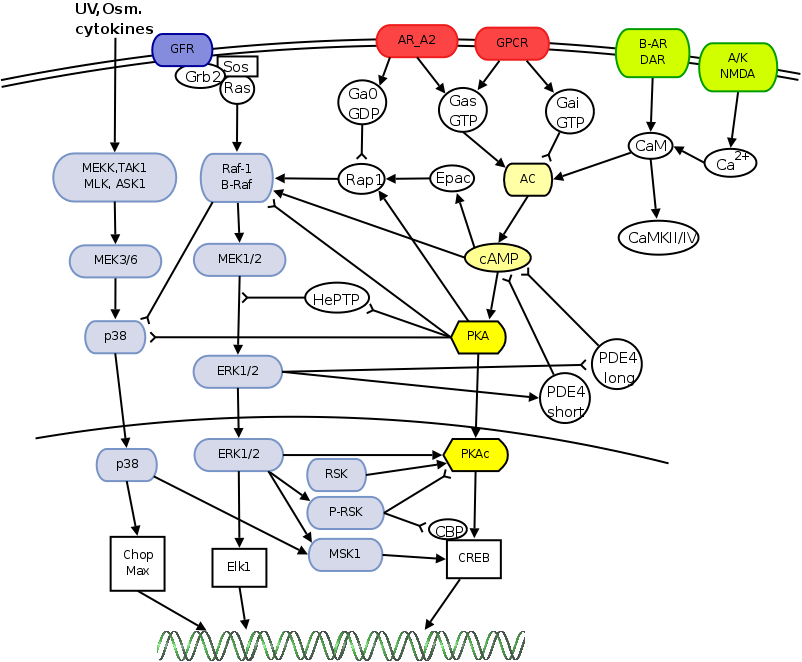
Figure 3.2: Possible ways for interactions between MAPK and PKA signalling pathways. The arrows indicate stimulation, the arrow ends inhibition. The Ga in Gas, Gai and Ga0 indicates the monomeric G® subunit coupled to GDP/GTP. See text for details. The figure is adapted from (65,83,300,299,151,334,127).
3.5.1 MAPK and PKA interactions
In unstimulated T- and B-cells, the haematopoetic tyrosine phosphatase (HePTP) binds ERK1/2 and p38 through its kinase interaction motif (KIM), removes their phosphate groups and thereby inactivates these MAPKs. Antigen stimulation of these lymphocytes increases the intracellular cAMP concentration, which activates PKA. PKA then phosphorylates HePTP on Ser23 in the KIM, thereby perturbing the binding site for MAPKs. This results in increased concentrations of active MAPKs and increases transcriptional activity of the c-fos promoter (318). In addition, active ERKs can phosphorylate RSKs, which elicits desensitisation of PKA for cAMP (see 3.5.2). It can also activate the short form of PDE4 which inhibits cAMP production, and inhibit the long form of PDE4, thereby stimulating cAMP synthesis and activation of PKA (151).
In the CA1 region of the hippocampus, NMDA receptors and Ampa/Kainate receptors can elicit a Ca2+ release in the cell. This stimulates calmodulin and AC, which increases PKA activity. PKA subsequently activates the MAPK pathway, which leads to increased CREB phosphorylation (299,394). The MAPK responsible for CREB phosphorylation was thought to be RSK2, however, recently Sindreu and colleagues proved that MSK1 phosphorylates CREB during Ca2+ -PKA-MAPK-induced fear conditioning (299,334). Another means to elicit MAPK activation via increased PKA activity occurs via the dopamine receptor and the ¯-adrenergic receptor (299).
In muscle cells, mAKAP is attached to the nuclear envelope by nesprin-1®. This complex gathers PKA, Epac, PDE4D3 and the Ca2+ release channel ryanodine receptor (RyR) together. PDE4D3 serves as a scaffold itself for MEK5 and ERK5. ERK5 phosphorylates PDE4D3, which results in decreased PDE activity. This implies that less cAMP gets degraded, which leads to increased PKA activity. Increased cAMP concentrations can also activate Epac and Rap1, which inhibit ERK5, thereby providing a negative feedback mechanism (93). The high cAMP concentrations also result in inhibition of ERK5 activity and transcriptional activation of c-Jun in HeLa and NIH3T3 cells. In contrast, low cAMP induces activation of ERK1/2 and ERK5 and leads to c-Fos and c-Jun transcriptional activation and cell proliferation (277).
Finally, during the process of integrin-mediated anchorage of cells, PKA influences the MAPK pathway as well. Cells deprived of anchorage, display increased PKA activity and reduced Raf-1 activity, whereas inhibition of PKA increases the activity of Raf-1. This opposite activity is the result of the phosphorylation of PAK1 by PKA. Phosphorylated PAK1 is inactive and cannot phosphorylate Raf-1 on Ser338 or MEK on Ser298. Thereby, Raf-1 remains inactive and interaction between Raf-1 and MEK is prevented (183).
3.5.2 MAPKAPK and PKA interactions
Crosstalk between the MAPK and PKA pathways occurs also in the case of RSK and PKA. In addition to sharing some substrates (CREB, BAD, ER81, GSK3, LKB1, Nur77), RSK and PKA can interact with each other directly. Inactive RSK can bind PKA RI in the presence or absence of AKAP. The interaction between inactive RSK and PKA RI attenuates the binding with PKA C subunits, which may sensitise PKA to cAMP or elicit cAMP-independent activation under the right circumstances. On the other hand, active RSK exclusively binds PKAC and promotes its binding to PKA RI, which desensitises PKA to activating stimuli (55,151).
The transcription factor ER81 is required for the formation of proper connections by sensory and motor neurons in the developing spinal cord. Its absence causes severe deficits in motor coordination. NTM and hormones activate PKA, which can phosphorylate ER81 on Ser191, Ser216 and Ser334. Phosphorylation of Ser334, reduced the DNA binding ability of ER81, but simultaneously enhanced its transactivation potential. This action may increase the gene expression from promoters that selectively bind ER81 with high affinity, and may not occur in promoters with low affinity for ER81. Similarly, GF-induced MAPK activation also results in phosphorylation of ER81 (Ser191 and Ser216) and CBP, increased gene expression (403).
Forskolin and ethanol both increase the activity of AC and lead to an increased cAMP concentration in the cell. cAMP then binds to RII¯C® and RI¯C¯ , which releases C® and C¯ respectively and increases their activity. The former catalytic subunit translocates to the nucleus where it phosphorylates CREB, the latter inhibits B-Raf in the classical MAPK pathway, and thereby relieves the CBP inhibition by p90RSK. This enhances CREB-mediated transcription in NG108-15 cells (65).
4. Initiation of transcription
The majority of signal transduction pathways end up in the nucleus where they influence the transcription of genes as part of a cellular response to a particular stimulus. This implies that gene expression patterns vary over time and that specific mechanisms regulate these patterns of transcription appropriately, a process called gene regulation. As Figure 4.1 illustrates, the regulation of gene expression occurs at several levels :
-
The level of accessibility represented by modifications in chromatin structure.
-
The level of transcriptional regulation, where the different elements that constitute a promoter drive gene transcription or not.
-
The level of post-transcriptional modifications where alteration of mRNA stability and availability of mRNA play a role.
-
The level of translation and post-translational regulation which determines the stability and activity of the newly formed protein.
Since the overwhelming majority of gene regulation occurs during initiation of gene transcription, this chapter will focus mainly on transcription-related processes (388).
|
|
||
|
|
|
|
|
|
||
Figure 4.1: The different levels at which gene regulation can occur: chromatin modifying factors allow for nucleosome displacements; histone modifying factors alter the affinity of histones for DNA; Mediator, cell specific transcription factors and negative co-regulators all influence transcription. In addition, distal regulatory elements can also contribute to the efficiency of transcriptional processes. Figure adapted from (301).
4.1 Gene regulation by modifying accessibility
To allow for several levels of compaction as well as for protection against DNases, positively charged DNA-binding proteins, histones, bind to (negatively charged) DNA. In this condensed state, the basal transcription machinery cannot reach the DNA, and by consequence, expression of the underlying genes is repressed (heterochromatin). In order to enable access, several factors can e.g. modify the affinity of histones for DNA or affect nucleosome location. In case of the former, the C-terminal part of a histone binds DNA whereas the N-terminal tails remain freely available for modifications like acetylation. Histone acetylases (HAT) couple acetyl groups to the Lys/Arg residues within the N-terminal tail of histones H3 or H4 and thereby introduce increased negative charges until it causes repulsion between the DNA and the histones. This makes the DNA available for binding by the basal transcription machinery and initiation of transcription (euchromatin). In contrast, histone deacetylases (HDAC) recruited by repressors, remove these negatively charged acetyl groups which attracts the histones to the DNA again and introduces compaction of DNA and repression of transcription (348,380).
Another means by which DNA can be made available for transcription, occurs by an ATP-dependent sliding of the nucleosomes along the DNA, or even involves transfer of the nucleosome to another chromosome. The mechanism of nucleosome displacement lies beyond the scope of this dissertation, but for more information, the reader may consult (139,402).
In GC-rich promoter regions (CpG islands), which often precede ubiquitously expressed housekeeping genes, methylation of cytosines represses activation of gene expression (216,354). This occurs by recognition of either several methylated cytosines or just one methylated CpG base pair by MeCP1 and MeCP2 respectively. MeCP2 can directly inhibit transcription by interacting with transcription complexes or it may become part of the Sin3 repressor complex, which possesses HDAC activity. Similar to CpG methylation, methylation of histones on Lys/Arg residues by histone N-methyl transferases also represses transcription (216,309).
Only few studies report a role for MAPKs in these processes. For example ERK1/2 and p38®/¯ can provoke changes in chromatin structure. This occurs most likely by their activation of MSK1/2 and RSK2. These kinases can phoshorylate the N-terminal tails of histones which allows further post-translational modifications (380), or e.g. RSK2 can interact with and regulate the HAT activity of CBP (242). As mentioned previously, ERK3 can phosphorylate histone H1 in vitro, but no in vivo evidence has been found yet (320).
4.2 Regions for control of gene transcription
In eukaryotic cells, certain DNA sequences (cis elements), to which protein complexes (trans elements) can bind, regulate the transcription of a gene. These sequences may lie close to the transcriptional start site (TSS) or further up- or downstream of the TSS. Together, all these sequences may be considered as a part of the promoter. On the other hand, a more restricted description of a promoter may only comprise the cis elements from which RNA polymerase II (RNPII) and the basal transcription machinery initiate gene transcription, the core promoter (216,388,139,197,222). These regions are summarised below and in Figure 4.2 :
|
|
||
|
|
|
|
|
|
||
Figure 4.2: Schematic representation of an active promoter. Abbreviations: NFR, Nucleosome Free Region; TSS, Transcriptional Start Site; BRE, TFIIB Recognition Element; MTE, Motif Ten Element; DPE, Downstream Promoter Element. The nucleosome region comprises approximately 150-200bp. The MTE has been found only in Drosophila, where it can function synergistically with the TATA-box or DPE to initiate transcription. The MTE requires the Inr for its functionality. The ``Proximal Promoter Elements'' and the ``Distal Promoter Elements'' are not indicated here. Figure adapted from (139).
-
The Core promoter: determines where RNPII binds the DNA and where it begins transcribing DNA into RNA. This region contains conserved sequences at fixed positions (-30 to +30; where +1 indicates the start site for transcription. Sequences downstream of the transcription start site are indicated by ``+'', whereas sequences upstream of the start site are indicated by ``-''.), which on their own provide for a low basal transcriptional activity (354,197) :
-
The Initiator (Inr), located near the start codon at position -2 to +4, consists of the pyrimidine-rich consensus sequence YYANWYY, where ``A'' represents the first base of the start codon. This element appears in 55% of the vertebrate promoters (both in TATA-less and TATA-containing promoters) and is recognised by TAF1 and TAF2 of TFIID (139).
-
The TATA box (or Goldberg-Hogness box) is located approximately 25 nucleotides upstream of the TSS in 10-16% of the vertebrate promoters. Its consensus sequence comprises TATAWAAR and is flanked by GC-rich sequences (139). The TATA-Binding Protein (TBP) of TFIID recognises and binds the TATA element where it functions as a binding site for RNPII. A minority of promoters lacks the TATA box, so called TATA-less promoters. These promoters often display a high content of CpG islands. In that case, TFIID binding occurs via TFIID Associated Factors (TAFs) that recognise the Initiator directly (216,388,139,197).
-
Heintzman and Ren also described a Downstream Promoter Element (DPE) on position +28 to +32 with consensus sequence RGWYN and the TFIIB Recognition Element (BRE) on position -37 to -32 with nucleotides SSRCGC. These elements appear in 48% and 12-62% respectively of the vertebrate promoters. Especially, TATA-less promoters possess DPEs. When recognised by TAF6 and TAF9 of TFIID, DPEs function as a downstream counterpart of the TATA-box to initiate transcription together with the Inr. Presence of both a TATA-box and DPE may influence enhancer interactions (129,139).
-
-
The Promoter Proximal Elements reside in the immediate neighbourhood of the core promoter (-30 to -200bp of the TSS). This region comprises transcription factor binding elements, whose binding aids or represses transcription (139,354,197,222) :
-
The CAAT box (``cat''-box) lies around position -75/-80 and consists of the GGCCAATCT consensus sequence. The element can be read in either direction and promotes the efficiency of gene transcription. Members of the CTF family (CP1/2; C/EBP, ACP) can bind to this element (216,197).
-
The GC box usually appears at -110/-90 but may lie as close as -40/-70bp upstream of the start codon. This GGGCGG-motif can occur as several repeats, to which the 105kDa transcription factor Sp1 binds. Sp1 binding can occur in either orientation and regardless of the presence of methyl groups on the GC box. Similar to the CAAT box, its binding increases transcriptional efficiency (216,197).
-
The Octamer consists of the 8bp sequence ATGCAAAG and it appears further upstream of the start codon. The transcription factor Oct1 can bind the octamer in non-lymphoid cells and thereby activate the transcription of H2B genes. Oct2 displays tissue-specific activation of Ig· light gene (216).
-
-
Distal Promoter Elements do not directly participate in RNPII binding in general, but influence the efficiency or rate of transcription. Their position varies from promoter to promoter, but they are usually located several 100kb up- or downstream from the TSS:
-
Enhancers function as sequences that increase transcription from a linked promoter, independent of their orientation or position. Similarly, silencers can elicit the repression of gene transcription (354,309). Recently, the presence of transcriptionally active general transcription factors (GTF) have been found near enhancers, which indicates that the presence of GTFs does not only remain limited to promoters. Even more, it may be possible that distal enhancers can be the first sites for GTF recruitment prior to activation of gene transcription (354).
-
Insulators are regulatory elements that establish independent domains of transcriptional activity by regulating which enhancers and repressors exert their effect on the transcription of a particular gene. More precisely, they can prevent enhancers from a specific promoter to activate an adjacent promoter or stop the spreading of heterochromatin which inactivates nearby gene transcription (216,222,309,206).
-
4.3 Initiation of transcription
When the DNA is accessible for the initiation of transcription, the TATA-binding protein (TBP) within TFIID binds the TATA-box. Subsequently, TFIIA and TFIIB join TFIID, after which RNPII with TFIIF, which contains helicase activity, bind the complex. Finally, TFIIE and TFIIH associate to form the pre-initiation complex (PIC). TFIIH contains ATPase, helicase and kinase activity. By means of its kinase activity, TFIIH can phosphorylate the C-Terminal Domain (CTD) of RNPII. This phosphorylation converts the PIC into a transcriptional elongation complex and induces the release of most of the TFII factors or GTFs. Moreover, the phosphorylated CTD of RNPII serves as docking site for e.g. capping enzyme (guanylyl transferase) (216,197,222,309).
|
|
|
|
|
|
|
|||
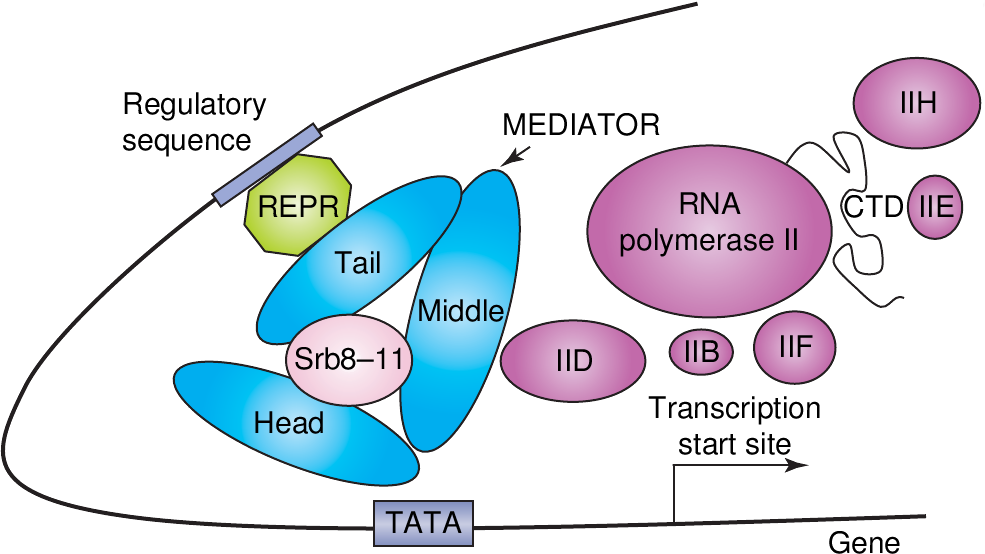 A.
A.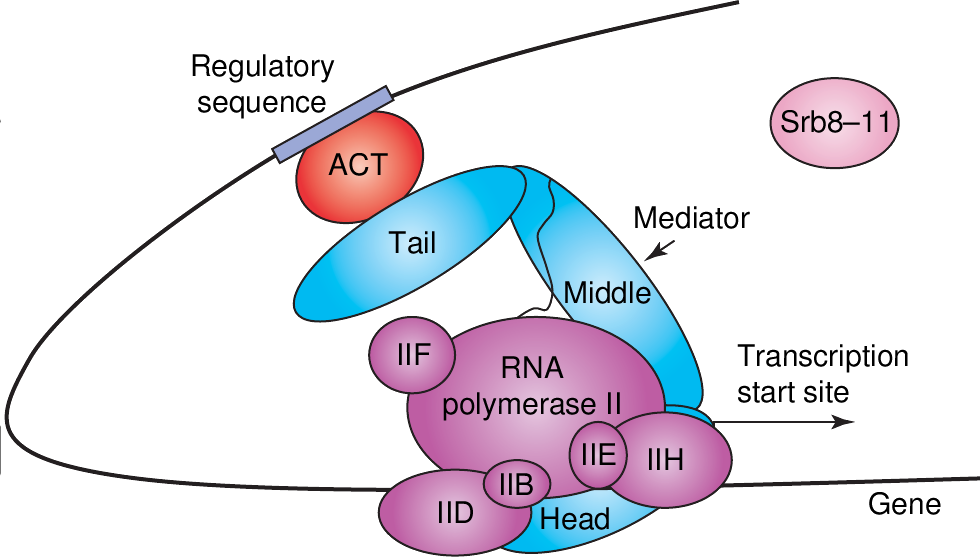 B.
B. Figure 4.3: The role of Mediator in initiation of transcription. Panel A: Repressor interactions (green) and binding of the cdk8/CycC subunit (pink) to Mediator (blue) prevent it from binding to RNPII and the basal transcription machinery (purple). Panel B: Activator (red) interactions allow Mediator to adopt an open configuration, which results in binding of RNPII and the basal transcription machinery. This initiates gene transcription (33).
As mentioned in section 4.2, the efficiency of transcription can be influenced by the involvement of activators and co-activators. One of the co-activators essential for basal transcription is called Mediator. Mediator has been isolated from Metazoan, Yeast, Drosophila, Mouse, Rat and Human cells. It consists of an elongated roughly conical 400Å complex of 20 to 30 subunits that together form a head, a middle part and a tail. As Figure 4.3A illustrates, binding of Mediator to a repressor results in a compact closed conformation and is accompanied by the presence of cdk8/CycC. This kinase complex may phosphorylate components of the basal transcription machinery, thereby introducing steric hindrance and repression of transcription. Similarly, phosphorylation of TFIIH decreases its kinase activity and results in reduced phosphorylation of the CTD of RNPII and thereby reduced transcription initiation (33).
Binding of Mediator to an activator shifts the middle part outward and opens up the tail. This open confirmation allows for the binding of RNPII to the head and middle part of Mediator (Figure 4.3B). Additionally, the tail of Mediator can now contact distant activators bound to specific regulatory sequences. This results in increased basal and gene-specific transcription by enhancing the phosphorylation of the CTD of RNPII via increased TFIIH kinase activity (63), or by facilitating the recruitment of TFIIB (19).
Similar to GTFs, hyperphosphorylation of the CTD leads to dissociation of Mediator from the PIC and may therefore not directly intervene during transcription elongation. Whether the function of Mediator remains limited to acting as a bridge between the activators and RNPII, or whether mediator also posseses other functions is still being investigated (33,27).
5. Transgenic mice
In fact there was only one species on the planet more intelligent than dolphins, and they spent a lot of their time in behavioural research laboratories running round inside wheels and conducting frighteningly elegant and subtle experiments on man. The fact that man completely misinterpreted this relationship was entirely according to these creature's plans. These creatures you call mice, you see, they are not quite as they appear. They are merely the protrusion into our dimension of vast hyperintelligent pan-dimensional beings. The whole business with the cheese and squeaking is just a front.. - D. Adams, The hitchhiker's guide to the galaxy
5.1 Introduction
Approximately 25 years ago, Palmiter and colleagues micro-injected a DNA construct bearing the promoter of the mouse metallothionein-I gene fused to the gene of rat growth hormone into the pronuclei of fertilised mouse eggs. One third of the progeny that developed from these genetically manipulated oocytes bore the transgene and became the first transgenic mice. This modification resulted in significantly larger transgenic mice compared to their littermate controls, thereby providing a first glance of the potential for animal research in the elucidation of the physiological role of a particular gene product (272).
Nowadays, there exist animal models for clinical conditions such as inflammation (416,358,128), cancer (172,304,114,387) and neurodegenerative disorders (108). In addition, there exist several animal models for particular families of protein kinases, including MAPKs. An overview of the most recent MAPK mouse models can be found in (120). This chapter summarises the technology available for the construction of transgenic mice, but will not address the rather extensive field of mouse phenotyping. For more information about phenotyping, the reader may consult (302,38,69).
5.2 Construction of transgenic mice
Depending on which aspect of a gene one wants to investigate, there exist three main directions :
-
Most functional studies of proteins involve the ``knockout'' of the gene that encodes a particular protein. A knockout mouse model lacks the exon of the gene that is critical for the expression of the gene product. The lack of such an exon results in the translation of an unstable protein or a non-functional protein and hence represents a ``loss of function''.
-
In another approach, one alters the function of the existing gene product by an amino acid substitution. This intervention is referred to as a ``knockin'' model and may lead to a ``gain-of-function'' or a ``loss-of-function'' for the gene product.
-
Finally, one may want to control when (e.g. during a particular developmental stage) or where (e.g. in a particular tissue) the gene of interest will be expressed (``conditional gene expression''). In practise, the introduction of a tetracycline responsive system or a tissue-specific promoter permits spatio-temporal expression of the transgene.
Each of these models demands a specific approach in terms of the construction of the transgenic vector, the introduction of the transgene into the organism, the expression of the transgene and last but certainly not least, the phenotyping. Except for the phenotyping, these concerns will be addressed in the following sections.
5.2.1 Transgene delivery
There exist three main ways to deliver a transgene into the cell, where each way requires a particular vector construct (summarised in Table 5.1):
Table 5.1: Specific vector requirements per method of gene delivery. Abbreviations: PGK, phosphoglycerate kinase promoter; NeoR , neomycin resistance; HSV, herpes simplex virus; TK, thymidine kinase; polII/III, polymerase II or III promoter; LTR, long terminal repeat; cPPT, central polypurine tract ; WPRE, woodchuck post-regulatory element; FLP, flipase; FRT, flipase recognition target; KanR , kanamycin resistance; TG, transgene. ``Transgene'' means here the introduced fragment.
|
|
Pronuclear injection |
Blastocyst injection |
Viral Delivery |
|
|
|||
|
Promoter |
Ubiquitous Tissue-specific |
PGK for NeoR and TK, or HSV for TK |
Pol II/III or 5' LTR |
|
Intron |
Desirable |
Genomic |
Desirable |
|
Transgene |
cDNA |
Genomic |
shRNA or cDNA |
|
PolyA |
Required |
No |
polyA or 3'LTR (cDNA) |
|
Regulatory elements |
No |
No |
cPPT and WPRE |
|
Recombination site |
No |
when Cre/loxP or FLP/FRT |
Yes |
|
Selection marker |
No |
Neor and TK |
NeoR, KanR,.. |
|
Reporter |
Desirable |
Desirable |
Desirable |
|
Site-specific |
No |
Yes |
Both |
|
Other |
removal of flanking plasmid sequences |
5' and 3' arms flanking the TG |
may need helper virus |
|
|
|||
5.2.1.1 Pronuclear injection
For pronuclear injection, the linearised and restriction enzyme digested vector is injected into one of the pronuclei of a fertilised mouse egg. During the subsequent cell divisions, the vector will integrate itself into the genome of the developing embryo. When integration occurs during the very first stages of cell divisions, the integrated vector will be present in the majority of the cells of the newly formed organism. If integration occurs during later stages, it will only be represented in a small amount of cells (e.g. in 10% of the germ cells). As such, it will be harder to discern which of these chimeric mice contain the transgene and which ones do not. Since integration occurs at random, several copies of the same vector often integrate as a concatamer at the same genomic site. In addition, the vector may also integrate at different places in the genome. This complicates interpretation of the effects observed in the mouse model later on, but can partially be overcome by determining the phenotype of several founder lines. If they are in agreement, the phenotype will most likely be due to the transgene and not to integration effects. Because non-homologous recombination occurs much more often than homologous recombination, pronuclear injection is a highly efficient process. Even more, due to few requirements for the vector, it remains a relatively straightforward process, and may yield pure strains of homozygous mice (222,227,308). In general, this technique is more often used in KI models and models that employ shRNA (see 5.2.1).
5.2.1.2 Blastocyst injection
For blastocyst injection, one first needs to select for the embryonic stem (ES) cells that have taken up the transgene. To this end, the linearised transgene-bearing vector will be transfected (often electroporated) into ES cells. Next, only ES cells that have integrated the vector into their genome and prove resistant to antibiotic resistance genes present on the vector, will survive. These positive ES clones will then be microinjected into the blastocysts, after which the blastocysts will be implanted into pseudo-pregnant mothers.
Vector development for blastocyst injection usually allows for genomic integration by homologous recombination and gives the advantage that the endogenous promoter drives the transgene expression. Hence, transgenic expression may occur under more natural circumstances. However, producing transgenic mice by means of blastocyst injection requires more time and is more labour intensive due to the additional demands for the vector and the selection of positive ES cell clones (227,222).
5.2.1.3 Viral delivery
Viral transgene delivery may provide a more natural way to generate transgenic mice. Adenovirus or adenovirus-associated viruses, and retroviruses are most widely used for these purposes. The former does not integrate into the hosts' genome, while the latter can integrate into the cellular genome to assure permanent transgenic expression. Retroviral integration requires safety measures to ensure that no live virus will be produced. These safety measures include the splitting of the viral vector into a transfer and integration vector, a structural and packaging vector, and envelope plasmids (274). Viral vectors can be transfected into early embryonic cells or injected into particular regions of the organism, like the brain, and are often used to deliver shRNA (148,108).
5.2.1.4 Knocking out a gene
5.2.1.4.1 Cre/loxP system
In the Cre/loxP system, cyclization recombinase (Cre) of bacteriophage P1 recognises the loxP DNA sequence and elicits DNA recombination. LoxP sites consist of an 8bp asymmetrical sequence interspersed between two 13bp palindromic sequences. Depending on the orientation of these sites, Cre either inverts the fragment in between the loxP sites (inverted loxP sites) or expels it as a circular fragment (direct repeats of lox P sites). If the genome and the vector each contain one loxP site, the vector will be inserted between the two loxP sites (26,227).
|
|
||
|
|
|
|
|
|
||
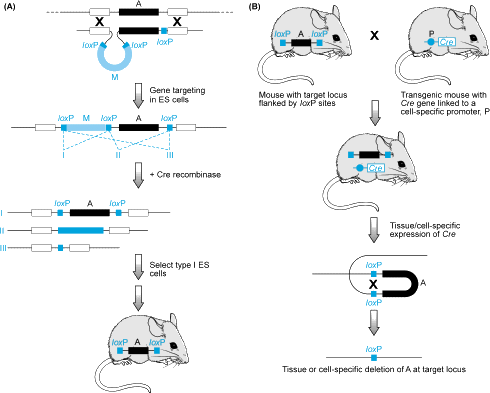
Figure 5.1: The Cre-loxP system. Panel A illustrates the homologous recombination of the targeting vector into the genomic DNA. Panel B illustrates how a gene is knocked out by crossing loxP and Cre mice (287).
Figure 5.1 indicates the process of generating a Cre/loxP mouse model. In order to generate such a model, one needs two mouse lines. One mouse line expresses the Cre recombinase (in all its tissues or in the tissue of choice; Figure 5.1 B), the other line contains the two loxP sites flanking the gene that needs to be deleted (``floxed''; Figure 5.1 A). Crossing of the two lines combines the loxP sites and the Cre recombinase, activates the recombination system and deletes the gene (311,227).
A system analogous to the Cre/loxP system is the FLP/FRT system, where Flipase (FLP from Yeast) initiates recombination by recognition of a 22bp flipase recognition target (FRT). The Cre/loxP system and the FLP/FRT system can be combined to generate conditional mouse models (53).
5.2.1.4.2 Ribozymes
Recently, Wallace et al introduced for the first time a more than 100kb sequence by means of the Cre/loxP system into the mouse genome. The fragment encompassed the floxed human orthologue of the mouse ®-globin gene, promoter and upstream regulatory sequences in an attempt to explain why the murine model of ®-thalassemia displayed a much milder phenotype compared to the human disease (393).
Ribozymes are catalytic RNA molecules or RNA enzymes that often catalyse their own cleavage from an RNA molecule. They were first introduced in mice by adenoviral-mediated transfer, where they targeted hepatic human growth hormone (hGH). The subsequent expression and action of the ribozymes ablated hGH expression for 96%, indicating another powerful tool to reduce gene expression (219). One of the most recent uses of ribozymes in MAPK mouse models was produced by Sun et al for the knockout of MK5/PRAK (see section 2.4.3) (351).
5.2.1.4.3 Short hairpin RNA (shRNA)
Like ribozymes, shRNA can only knockdown gene expression. This may be of interest when lower levels of expression are required, as opposed to complete KO. Viral vectors or pronuclear injection can both deliver shRNA either to ES cells for permanent reduction or to the tissue of choice for temporary expression (406,210). The production of shRNA may involve type II or type III RNA polymerase promoters, but depending on the choice of promoter, gene silencing can occur in two ways. Type III promoters elicit the traditional RNAi activation, whereas type II promoters first induce miRNA formation before stimulating the RNAi machinery (406,68,210,269). Multiple shRNA mouse models have been constructed and the reader is referred to (406,378,269) for more information.
5.2.1.5 Temporal expression
|
|
|||
|
|
|
|
|
|
|
|||
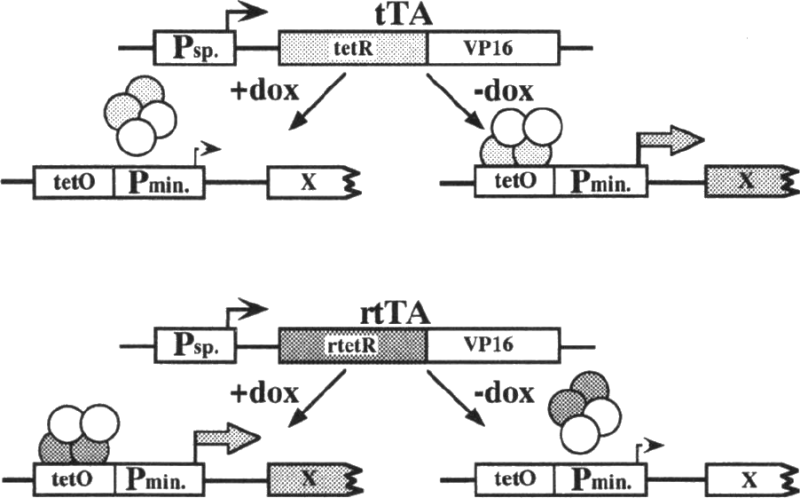 A.
A.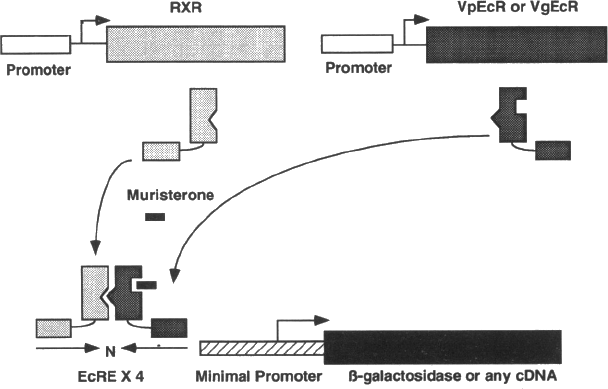 B.
B. Figure 5.2: Two inducible systems for the expression of a transgene. Panel A illustrates the TetOn/Off system, where doxycycline regulates transcriptional activity (194). Panel B illustrates the ecdysone system, where ecdysone binding regulates gene expression (267). For more details see text.
Providing one knows how long it takes for the inducing agent to turn the system on or off and how long the transgenic protein and its mRNA remain stable, temporal expression systems seem very promising. They allow the researcher to carefully tune the expression of their transgene in a ubiquitous or tissue-specific way (227). Figure 5.2 illustrates the widely used TetOn/Off system and the ecdysone system.
5.2.1.5.1 TetOn/TetOff system
The basis for the TetOn/TetOff temporal expression system lies in the tetracycline (Tc) transactivator (tTA) and a modified CMV promoter. The tTA consists of a fusion between the tetracycline repressor (TetR) of the Tn10 resistance operon from E. coli and the transactivation domain of VP16 from Herpes Simplex Virus (HSV). The promoter region to which this fusion protein binds consists of a minimal human CMV promoter which possesses seven cognate operator sequences (TetO). In the absence of Tc, tTA binds TetO and activates, in stead of represses, gene transcription. This represents the principle of the TetOff system. The TetOn system consists of a mutated tTA, rtTA, which only differs from tTA by four amino acids. This difference results in a fusion protein that requires the presence of Tc in order to bind to TetO to activate gene expression. The latter system displays increased kinetics and does not require continuous administration of Tc/doxycycline in the drinking water of mice (194).
Mayford and coworkers nicely illustrated the potential of this technique in a transgenic model of a calcium-independent form of CaMKII, where the mice behaved normally during repressed transgene expression, but displayed absence of hippocampal LTP, impaired fear conditioning and deficits in spatial memory during transgene expression. When the expression of the transgene was switched off again, the abnormal phenotype disappeared (235).
5.2.1.5.2 Ecdysone system
The steroid hormone 20-OH-ecdysone binds to the ecdysone receptor in insect cells, after which it dimerises with USP and triggers metabolic changes. In order to increase the sensitivity and response to an ecdysone stimulus for application in mammalian cells, No et al replaced the ecdysone receptor's TAD by that of the HSV VP16 protein. In addition, they introduced the mammalian homologue for USP, RXR, as its heterodimerisation partner, which increased the expression of the TG. Because of the lack of ecdysone in mammalian cells and its low toxicity, the ecdysone system may provide an alternative for the toxic TetOff system. Furthermore, it allows for a faster distribution and clearance of the ecdysone inducer. However, currently, the ecdysone system is far less popular than the Tet-inducible system (267).
5.2.1.6 Tissue specificity
Numerous examples of tissue-specific expression of a transgene have been described in the literature. For example, a Cre recombinase under the control of a nestin promoter, limits the expression of Cre to neurons and glial cells in mice. Crossbreeding of these mice with a mouse line that contains a floxed gene of interest allows for selective deletion of the gene only in neurons and glial cells (378). Several tissue-specific Cre mice have been developed. Recently, Cre expression was placed under the control of a BAC containing the foxl1 promoter and allowed expression of Cre only in the gut mesoderm (311).
There exist also several mouse models that overexpress the transgene in a tissue-specific way. For example, the cDNA of preprogalanin under the control of the dopamine ¯-hydroxylase promoter expressed the inhibitory neuropeptide galanin primarily in noradrenergic neurons. In this way, researchers examined the role of galanin in Alzheimer disease-like neurochemical and cognitive impairments in the basal forebrain (345), morphine withdrawal in the locus coeruleus (415) and its role as an endogenous anti-convulsant in the hippocampus (236).
An example of non-neuronal tissue-specific transgene expression comprises a mouse model of constitutive active MKK6, the upstream activator of p38. In this model, the Collagen type II ®-1 (Col2a1) promoter targeted MKK6E/E cDNA and LacZ mainly to chondrocytes, which resulted in a dwarf phenotype with deficits in chondrocyte proliferation and differentiation (418).
5.2.1.6.1 Tetraploid rescue
Tetraploid rescue can be considered as a special case of tissue-specific transgene expression. With this technique, one circumvents potential embryonic lethality upon the deletion of a particular gene by using tetraploid WT cells. These WT cells can be produced by electrofusion of diploid WT embryos at the two-cell stage. When mixed with normal diploid ES cells that contain the KO for a particular gene, tetraploid cells will only contribute to the development of extra-embryonic tissues, and not intervene in the development of the embryo itself (164). For example, MAPK KO models of MEK1, p38® and ERK2 all produced embryonic lethality, which could be circumvented by tetraploid rescue (134,253,357,2,31).
5.2.2 Transgene expression
Transgenic expression or endogenous gene knockout can be detected by several methods. For example, failure to detect the endogenous protein in a western blot by using specific antibodies confirms the KO. On the other hand, western blot in combination with transgene-specific antibodies can detect overexpressed or modified transgenic proteins and thereby distinguish it from the endogenous one. To that end, the transgenic construct is often coupled to tags, like the Hemagglutinin (HA) tag derived from the HA-protein from influenza virus or enhanced green fluorescent protein (EGFP). Similarly, one can include the lacZ gene which encodes ¯-galactosidase. This enzyme converts lactose into glucose and galactose and cleaves the colourless X-gal into galactose and an insoluble blue product. This visualises transgene expression or deletion in particular tissues.
A recent technological development uses quantum dots for fluorescent detection of DNA or proteins. In Fluorescent In Situ Hybridisation (FISH), streptavidin-linked quantum dots (as crystals of (CdSe)ZnS) bind to biotin-16-dUTP labelled DNA. Depending on the size of the quantum dot, the emitted fluorescence lies more towards the red or the blue spectrum (407). Quantum dots may also be used to reduce the unspecific effects of Fluorescence Resonance Energy Transfer (FRET). External excitation light causes excitation of the fluorophore and emission of fluorescent light. However, despite the fluorescence emitted by the fluorophore, the external light source also produces unspecific fluorescence due to the presence of hormones and flavines. In Bioluminescence Resonance Energy Transfer (BRET), the quantum dots receive energy from the light emission of an associated luciferase. This results in a highly specific emission near infrared wavelengths and low background or auto-fluorescence. The use of quantum dots still needs improvement in terms of coating for solubility, questions of immunogenicity and clearance from the body. Nevertheless, in nude mice, quantum dots can be detected at a depth of 3mm (119,113,106,51).
6 Aims of the study
The two independent research groups, that isolated MK5 in 1998, described its expression pattern and some of its biolochemical properties e.g. the conserved phosphorylation site, some in vitro substrates and external stimuli (262,264). When the work of this dissertation was initiated in 2004, the knowledge about MK5 remained limited: others and we had studied the subcellular localisation, showing that MK5 contains a functional NLS and NES (324,261), while another group demonstrated no obvious phenotype in MK5 deficient mice (328). Neither upstream activators nor genuine substrates were known at the time. Therefore, the overall aims of this study were to elucidate the functions of MK5 and to increase our knowledge about this kinase. More specifically, the aims were:
-
to generate a transgenic mouse line that expressed the constitutive active MK5L337A mutant,
-
to examine the phenotype of this mouse line to gain insight into the biological role of MK5,
-
to investigate a role for MK5 in cell differentiation induced by the cAMP/PKA signalling pathway,
-
to explore the involvement of MK5 on cellular gene expression by comparing the transcriptome in control cells with cells expressing a constitutive active MK5L337A mutant,
-
to study the expression pattern of MK5 in detail,
-
to explore whether transcription of the mk5 gene was inducible by specific stimuli,
-
to identify the promoter elements necessary to mediate stimulus-induced transcription of the mk5 gene.
7 Summary of papers
Paper 1 :
Modulation of F-actin rearrangements by the cyclic AMP/PKA pathway is
mediated by MAPKAP Kinase 5 and requires PKA-induced nuclear export
of MK5. Nancy Gerits , Theresa Mikalsen
, Sergiy Kostenko, Alexey Shiryaev, Mona Johannessen and Ugo
Moens. Journal of Biological Chemistry
October 2007. In Press
, Theresa Mikalsen
, Sergiy Kostenko, Alexey Shiryaev, Mona Johannessen and Ugo
Moens. Journal of Biological Chemistry
October 2007. In Press
In this paper, we show that the catalytic subunit of PKA (PKAC® ) directly interacts with the MAPK-activated protein kinase MK5, but not with its family member MK2. This interaction between PKAC® and MK5 leads to an increased phosphorylation of MK5, as determined by in vitro kinase assays and simultaneously boosts MK5 activity levels (as determined by in vivo kinase assays). In contrast to inactive MK5 which resides mainly in the nucleus, the activated MK5 translocates to the cytoplasm. Both FSK stimulation or presence of PKAC® can elicit such a localisation and depends on the activity of both kinases, since neither a kinase dead MK5 nor a kinase dead PKAC® can elicit this nuclear export. Similarly, the location of both kinases plays a vital role in this process because nuclear exclusion of PKAC® does not, while exclusively nuclear PKAC® can induce MK5 nuclear export. Next, we found that FSK-induced MK5 activation and translocation induces F-actin rearrangements. Both processes are linked because the time points at which translocation occurs, overlap with those during which F-actin rearrangements take place. In addition, we found that siRNA against MK5, inactive MK5 or inhibition of nuclear entry by PKAC® prevented these F-actin rearrangements. From these data, we conclude that FSK/PKA-induced temporary nuclear export of MK5 modulates F-actin rearrangements, a process involved in processes like cell differentiation and cell motility.
Go to Paper 1
Paper 2 :Intensity Dependent Confidence Intervals on Microarray Measurements of Differentially Expressed Genes: A case study of the effect of MK5, FKRP and TAF4 on the transcriptome. Werner Van Belle, Nancy Gerits, Kirsti Jakobsen, Vigdis Brox, Marijke Van Ghelue, Ugo Moens. Gene regulation and Systems Biology, 2007;1:57-72
In order to determine whether MK5 could affect gene expression, we constructed a stable and inducible cell line for MK5L337A. We performed microarray experiments from RNA isolated from cells where expression of MK5L337A was turned on and off. In addition, we included a microarray slide with two exactly the same samples to be able to compensate for the measurement errors. This enabled us to devise a new way to analyse microarray data based on intensity-dependent confidence intervals. To test whether our method worked on other samples, we compared gene expression before and after knockdown of FKRP and TAF4, and validated and confirmed the microarray results with qPCR. We also compared our method with the existing techniques and found that our method is more accurate.
Go to Paper
2
Paper 3 :Transgenic mice expressing constitutive active MAPKAPK5 display gender-dependent differences in exploration and activity. Nancy Gerits, Werner Van Belle and Ugo Moens. Behavioral and Brain Functions, 2007;3:58
Since knowledge about the biological function of MK5 remains limited, we constructed a mouse model that expresses a constitutive active variant of MK5, to which we referred as MK5L337A mice. These mice were viable and fertile and displayed no gross abnormalities. During background breeding, we performed the primary SHIRPA protocol on transgene positive mice and their littermates. From these observations, we hypothesised a difference in anxiety related behaviour. As such we performed two validated anxiety tests, namely the elevated plus maze (EPM) and the light-dark box (LD) test. These tests revealed increased exploration of female transgenic mice (FTG) mice compared to female non-transgenic mice (FNTG) mice as measured by the amount of head dips and time spent on the open arm of the EPM. We also found increased locomoter activity of male transgenic mice (MTG) mice compared to male non-transgenic (MNTG) mice, as measured by the amount of transitions between the light and dark compartment in the light dark box for MTG mice compared to MNTG littermates. In addition, our image analysis method also allowed us to detect that FTG explored further into the open arm on the EPM compared to the other groups, and that these mice are less active in the closed arm compared to FNTG littermates. In contrast, MTG mice are more active than MNTG mice in the closed arm of the EPM. Since differences in the levels of neurotransmitters can influence anxiety-related processes as well as locomotion, this may indicate for the first time a role for a MAPKAPK in neurotransmitter regulation.
Go to Paper 3
Paper 4 : Transcriptional regulation and cell-specific expression of the mitogen-activated protein kinase-activated protein kinase MK5. Nancy Gerits, Alexey Shiryaev, Sergiy Kostenko, Helle Klenow, Olga Shiryaeva, Mona Johannessen and Ugo Moens. Manuscript.
In this manuscript, we compared the MK5 transcript levels of different tissues by performing tissue arrays. From these data we can conclude that MK5 expression occurs in a spatio-temporal way. Therefore, we wanted to investigate which stimuli could regulate the expression of MK5 transcripts from the MK5 promoter. To this end, we stimulated PC12 cells with KCl, sorbitol, retinoic acid, vanadate, FSK, lipopolysaccharide, spermine NONOate, serum, TPA (a phorbol ester) and induced a temperature shock. Of all the stimuli we tested, only FSK and heat shock elevated the MK5 transcript levels, whereas FSK did not alter MK5 its protein levels. Analysis of the promoter regions of the murine, rat and human MK5 promoters revealed that each of these promoters contained non-consensus CRE elements. First, we cloned an 850bp fragment of the human promoter, which contained a CRE element, into a luciferase reporter plasmid. Although CREB is able to specifically bind to this CRE element in vitro, this reporter plasmid remained unresponsive to FSK upon FSK stimulation. On the other hand, mutation of this CRE element reduced basal transcription levels 10-fold. Currently, we cannot explain the discrepancy between MK5 transcript levels and protein levels.
Go to Paper 4
II. Discussion
Discussion
It is like a finger that points to the moon. Don't look at the finger, or you'll miss all the heavenly glory.. – Enter the Dragon 1973
In this dissertation, different approaches were used to find out more about the biological function of the MAPKAPK, MK5 or PRAK. In what follows, I will use the bottom-up approach to discuss our experiments, approaches and findings, starting at the DNA level, through the protein level to interactions and physiological processes.
1. Regulation of the MK5 promoter
How a promoter responds to particular stimuli depends on the cell type and the presence of particular promoter elements (see chapter 4). Previously, a study in the rat tyrosine hydroxylase and other promoter regions, found that if CRE elements resided further than 250bp upstream from the TATA-box, they were less responsive to FSK than those that occurred in close proximity to the core promoter (180,64,373). In Paper 4, we found that the CRE element of the hMK5-LUC resided outside the 250bp region, which may explain the observed lack of cAMP response. On the other hand, this could also be attributed to the lack of a TATA-box in the MK5 promoter, which also results in reduced responses to FSK stimulation (64). However, since MK5 transcript levels from the endogenous promoter increase after FSK treatment, this seems unlikely. A more likely explanation may lie in the absence of upstream regulatory elements which are required for FSK-induced increases in MK5 mRNA.
Increased amounts of transcript can result from either increased transcription or from the stabilisation of these mRNA molecules. Previous studies have shown that cAMP can affect the stability of mRNA (180). We addressed the former in Paper 4, where we found that FSK treatment enhanced MK5 transcription. However, the increased amount of MK5 transcript did not result in augmented MK5 protein levels. This may indicate that either FSK increased ubiquitin-mediated proteasome degradation or that MK5 transcripts are destabilised by RNA binding proteins. These proteins bind to AU-rich elements, or ARE sequences and thereby affect the stability of the bound transcript (22). However, since the MK5 mRNA sequence, like other MAPKAPKs, does not contain ARE-like consensus sequences (21), it seems unlikely that ARE-binding proteins are involved. This contrasts to ERK3, whose sequence contains Class I ARE sequences, prototypically found in mRNA that encodes transcription factors or cell cycle regulatory proteins. This is consistent with predicted functions for ERK3 and its interaction partner cyclin D3.
In Paper 4, we also applied stimuli that are known to elicit MAPK activation and which converge to motifs present in the MK5 promoter to find out to which stimuli the MK5 promoter can respond. However, except for FSK and temperature shock, none of these stimuli could augment MK5 transcript or even protein levels. This resembles the lack of response from another MAPK, ERK4, which cannot be activated by classical MAPK stimuli (426). Since ERK4 and MK5 interact, it may be possible that they share a common upstream signal. Even more, ERK4 transcript could become upregulated by FSK, however such a hypothesis has not been tested yet. The lack of response may also be explained by the fact that different cell types respond differently to particular stimuli, and that in this case, PC12 cells could be less suited to assess the responses to the tested compounds. Therefore, it will be interesting to determine the MK5 promoter activity in different cell types treated with different stimuli.
2. MK5 protein structure
2.1 MK5 structure
Currently, the 3D structure of MK5 remains unresolved. In order to understand better how MK5 is folded we tried to construct an in silico model of MK5 via threading, but due to time issues, we could not finish a suitable model. Indirectly, we obtained evidence that the K51 residue seems to play an important role during folding of the protein: in contrast to all mutant MK5 fusion proteins which we could express and purify from bacteria, the K51E MK5 mutant could only be extracted from an eukaryotic reticulocyte lysate based in vitro transcription/translation system. This may indicate that specific eukaryotic factors are required for the proper folding of MK5K51E.
2.2 MK5 interaction
In vitro kinase assays of MK5 with several kinases resulted only in phosphorylation of the ``un-tagged'' MK5, indicating that tags may hinder studies that aim to find suitable interaction partners for MK5. This implies that endogenous protein forms should be used along side as a control.
When exploring the PKA-MK5 interaction, we screened for the involvement of putative PKA phosphorylation sites (T14, S43, S115 and S348), for which we used the NetPhosp Server (36), or the K51E and T182A MK5 mutants. Since subsequent studies did not reveal any clear involvement of either of these residues from in vitro kinase assays and the confocal microscopy experiments, we constructed double, triple and quadriple mutants. We are in the process of assessing if they affect the MK5-PKA interaction and the PKA-induced nuclear export of MK5. Moreover, we will investigate the effect of these mutations on MK5 its kinase activity. A recent resubmission of the MK5 sequence to the same server, only found S115 as a putative PKA phosphorylation site, which may in part explain our -lack of- findings. However, this does not necessarily mean that PKA cannot phosphorylate other residues in non-consensus PKA phosphorylation sites on MK5. Indeed, most of the PKA substrates lack this ``consensus'' phosphorylation site. This may also explain why the anti-phospho PKA-substrate antibodies in Paper 1 did not work when we tried to detect increased phosphorylation levels of MK5 by PKA.
3. MK5/ERK3 versus LKB1/STRAD ?
Schumacher et al (320) mentioned that activation and nuclear export of MK5 via ERK3 resembled the activation and nuclear export of LKB1 via STRAD (17). In fact, it seems this analogy goes even further than activation and nuclear export. For example, both LKB1 and MK5 interact with PKA and become activated after FSK stimulation. The former kinase becomes stabilised by FSK and affects axonal guidance (327). However, LKB1 does not undergo PKA-induced relocation (62), in contrast to MK5, where PKA both increases MK5 activity and induces nuclear export, which are required for F-actin rearrangements (Paper 1). Activation of LKB1 can also rearrange the actin cytoskeleton. In this case it induces the formation of an apical brush border. Loss of LKB1 causes cellular transformation through disruption of epithelial polarity (18). A role for MK5 in cellular polarity as the one described for LKB1 remains to be elucidated, but loss of MK5 can also induce cellular transformation through its lack of p53 phosphorylation, which exerts a pro-apoptotic effect on the cell. Indeed, a tumour suppressive role that involves activation of the Ras-p38-MK5-p53 pathway has been illustrated in MK5 KO mice that display enhanced skin pappillomas upon DMBA treatment (351). Similarly, LKB1 functions as a tumour suppressor protein that becomes activated by Ras and that phosphorylates p53 (188,176). LKB1 mice+/- develop polyps and hepatocellular carcinomas, whereas LKB1-/- in a C57BL/6 background die in utero around E8.5-E9.5 (161). Embryonic lethality of MK5 KO mice in a C57BL/6 background has also been reported. Here, pups die at E11 due to incomplete penetrance, which may reveal a role for MK5 in cell migration during embryonic development (320). At that time point during development, protein levels of the unstable ERK3 peak. These increased protein levels may be attributed to the presence of its stabilising interaction partner MK5. A more speculative link between MK5 and LKB1 may lie in the cell cycle, where LKB1 expression induces G1 cell cycle arrest (371). Since MK5 can stabilise ERK3, it may represent the physiological means that helps to induce G1 cell cycle arrest during increased ERK3 expression (184). Finally, AMP-activated kinases may provide an even more speculative resemblance between MK5 and LKB1, since LKB1 can phosphorylate and thereby activate AMP-activated kinase (400). Our protein interaction map indicates that constitutive active MK5 also affects AMP-activated kinases, but whether it does so or what the underlying mechanism might be, remains unknown (unpublished data based on Paper 2).
4. F-actin rearrangements
Since filamentous actin (F-actin) constitutes a component of the cytoskeleton and the cytoskeleton determines the structure and flexibility of a cell, F-actin rearrangements may underlie the basis of cell migration, neuronal flexibility and differentiation (see Figure 5.3).
|
|
||
|
|
|
|
|
|
||
Figure 5.3: Overview of putative MK5 functions. For details see text and ``Future perspectives''.
4.1 Cell migration
During development, many cells migrate from one region to their destination following gradients of signalling molecules. Typically, disturbances in such migration result in embryonic lethality as illustrated by mice deficient in the Met tyrosine kinase receptor. In absence of Met, scatter factor, the ligand of Met, cannot bind to its receptor and thereby not induce migration of muscle progenitor cells to the extremities (30). The MK5 KO mice in a C57BL/6 background reported by Schumacher et al also displayed partial embryonic lethality with incomplete penetrance. The authors suggested that defects in cell motility could lie at the basis of this defect (320,117). In addition, we found in Paper 1 that MK5 plays a role in F-actin rearrangements, which may indeed indicate a role for MK5 in cell migration.
Another example whereby signalling affects the cell motility occurs via monomeric Rho GTPases (mentioned in 1.2.2). For example, RhoA gradients in the neuron accumulate from the soma towards the leading edge of the neuron and determine the direction for cell migration. The BTB domain in Rho GTPases allows them to influence migration by binding to and organising actin filaments (29). Aside from a proven role in FSK-induced F-actin rearrangements (Paper 1), overexpression of MK5L337A in our inducible PC12 TetOff cell line caused the upregulation of Rho GTPase-related genes and several other cytoskeleton-related proteins, thereby solidifying a role for MK5 in cytoskeletal rearrangements (unpublished findings from Paper 2). Another MAPKAPK, MK2 has also been implicated in cytoskeletal rearrangements, where MK2 can phosphorylate Hsp25 and thereby allow for the addition of new G-actin monomers onto the barbed end of actin (202).
4.2 Neuronal flexibility
Neuronal flexibility in learning processes requires structural reshaping of neurons and hence involves the cytoskeleton and F-actin. No reports indicated a role for MK5 in learning and memory processes yet, but many reports have shown that PKA as well as other MAPKs can play a role in such processes. For example, KO of ERK1 increases striatal-mediated learning and memory and facilitates synaptic plasticity in the striatum (237). Furthermore, the PKA and MAPK signalling pathway are necessary for memory retrieval and extinction in the CA1 region of the hippocampus of fear-conditioned rats (313,353). In addition, the MAPKAPK, MSK1 also signals in concert with the Ca2+-stimulated adenylyl cyclase activity during fear conditioning to activate CREB (334).
In Paper 4, we find that CREB can bind to the MK5 promoter and that MK5 transcript transiently elevates, with highest levels at 30 minutes to 1 hour. CREB phosphorylation after memory training occurs in two waves (225,377,163): the first wave of phosphorylation by PKA occurs after 30 minutes, the second wave 3 to 6 hours after training. One could speculate that these increases of MK5 transcript are connected to early events in memory formation. In that regard, it could be interesting to subject our MK5L337A mice (Paper 3) to learning and memory tasks.
4.3 Differentiation
F-actin rearrangements also occur during differentiation of PC12 cells after NGF and dbcAMP treatment. Treatment of PC12 cells with NGF induces increased stabilisation and hence accumulation of ERK3, which is accompanied by increases in MK5 activity in a p38-independent way (325). However, in contrast to RSK1 (333), the expression of constitutive active MK5 in our inducible PC12 TetOff cell line (Paper 2) alone is not sufficient for differentiation, since we observe no differences in neurite outgrowth with or without this variant. On the other hand, these differences may be explained by the subcellular localisation, since the L337A mutant resides exclusively in the nucleus while dbcAMP (and maybe NGF) provoke nuclear export of MK5. Transient or stable expression of an active MK5 to which a functional NES is fused (like the MK5L337A-NES in Paper 1) could allow us to test this assumption. Given the transient alterations in the F-actin cytoskeleton, it may be possible that MK5 serves as a switch to induce particular cellular responses where the cell context (e.g. activation of FSK-PKA-MK5 or NGF-ERK3-MK5) directs the outcome of the response e.g. in a ``motility direction'' or in a ``differentiation direction''.
5. MK5L337A transgenic mice
Although at the time point of construction, the approach we followed was the best compromise, we could have approached the construction of these mice in a different way. For example, the NTS offered us only blastocyst injection, and it provided us with fertilised eggs in a C57BL/6XCBA background. This prevented us from using site-directed knockin and obliged us to breed the mice into the full C57BL/6 background. On the other hand, the development of a vector for blastocyst injection and the screening of positive ES cells would require a longer time for construction than we needed now, and would certainly not have yielded results at this time point yet.
Furthermore, we experienced difficulties during the detection of transgenic MK5 protein, which we could have circumvented had we left the ¯-galactosidase fragment in the CMV-plasmid we used as a vector. In a similar way, we could not trace back where in the mouse genome our transgene integrated. We used several strategies to find the integration site of the transgene including Southern blot, FISH, outward sequencing and fusion of linkers to the restriction-enzyme digested genomic DNA fragments, which were subsequently cloned, amplified and sequenced. Although we picked up some positive clones containing the transgenic MK5 sequences, there was no uniform answer as to where the transgene was integrated and hence, we could not determine the transgenic integration site with certainty.
Consequently, this implies that in order to be sure that the effects we see are due to the transgene and not arise because of side effects due to the site of integration, we need to phenotype several mouse lines. Our mouse lines A and B are due for cyropreservation, however our line C died during background breeding. One of the main reasons probably lies in the background breeding itself rather than due to the transgene, since line A and line B both express the transgenic MK5 transcript and can be bred without problems. Histochemistry of some pups in the F3 generation of line C revealed that some of the mice failed to inspire air at the moment of birth, as shown by a lack of air in the lungs.
Next, we planned on phenotyping our mice at the German Mouse Clinic in Munich, however, the sentinel animals which resided in the same room as our transgenic animals at the animal facility contracted the mouse pneumonia virus. Because of the risk for infection, we could not send our mice for phenotyping, even though our mice appeared negative during all screening steps. This meant that either we had to abrogate the entire project due to extra time and money issues to expand a colony once again, or to try to perform an experiment by ourselves with the mice we had. Since we performed the SHIRPA protocol on all the animals during background breeding, we had some indication as to which test would yield the most information. We decided to test our mice on the EPM and LD since these tests are non-invasive, easy to perform and require no conditioning and consequently no additional approval from an ethical committee.
As discussed in Paper 3, we found gender-dependent differences in exploration and locomotor activity of the MK5L337A mice. Considering that the L337A mutation disrupts the NES signal of MK5 and can not induce F-actin rearrangements because it does not leave the nucleus (Paper 1), the MK5 transgene in our mice could not interact with its cytoplasmic partners and hence we may have detected only a subtle variant of the ``real'' phenotype. As such another mouse model that combines both an activating mutation and an extra intact NES signal in the endogenous MK5 sequence may allow for a more pronounced phenotype.
Conclusion
In this study we set out to find out more about the physiological role of the poorly characterised MAPKAPK, MK5. Our studies show that PKA interacts with, phosphorylates and activates MK5. This interaction requires the activity of both kinases and leads to nuclear export of MK5, which is required for F-actin rearrangements. In addition, we find that expression of MK5L337A alters gene expression of genes involved in metabolism, transcription and translational processes, cytoskeletal organisation and signalling. We also constructed a mouse model that enabled us to detect gender-dependent differences in exploration and locomotor activity. Here, female transgenic mice proved less anxious as indicated by their increased exploration, whereas male transgenic mice displayed enhanced activity. Finally, the mk5 gene seems to be regulated in a spatio-temporal manner, where traditional MAPK stimuli can not elicit elevated transcript or protein levels in PC12 cells. However, we do see a difference in transcript levels upon heat shock and FSK treatment. Further studies are required to elucidate all the functions of MK5.
9 Future Perspectives
1. Future experiments
The present study leaves several questions
unanswered. Following suggestions might help elucidate those :
-
A role for MK5 in F-actin rearrangements related to cell migration : Migration assays of PC12 cells in response to FSK with cell transfected with different mutants of MK5, MK5 siRNA or PKI expression plasmids in order to further assess the consequences of F-actin rearrangements.
-
Transcriptional regulation of the MK5 promoter : A comparison in responses after the stimuli tested in Paper 4 between the mouse, rat and human MK5 promoter in their respective cell lines. In addition, cell lines other than neuronal cell lineages might be tested as well.
-
Verification of the microarray data from the stable inducible MK5L337A PC12 cell line by qPCR.
-
Transgenic mice :
-
Line A of the MK5L337A can be tested on the EPM and in the LD, and/or phenotyped.
-
Considering money and ethical approval, one could construct a mouse model with a targeted mutation of L337A into the endogenous MK5 sequence combined with an additional NES and a reporter. Limited tissue expression of such a transgene to brain, heart and skeletal muscle would be very interesting, as these tissues display high expression levels of MK5. Since both ERK3 and MK5 KO mice display embryonic lethality, it could also be interesting to construct a conditional knockout model.
2. Hypothesis
A role for MK5 in vesicle transport ?
From the data obtained in the presented papers and from unpublished results, I would like to hypothesise a role for MK5 in vesicle transport.
Context
Neuronal signalling involves the transmission of signals by propagating an electrical discharge along their membrane, the action potential (AP), and by the synthesis of neurotransmitters (NTM) that are released in the synapses between neurons.
-
AP : This self-propagating depolarisation and repolarisation process requires the alteration of ion fluxes over neurons' membrane and involves voltage gated ion channels in the cell membrane. The change in membrane potential opens up the voltage-gated K+ channels that allow the outflux of K+ ions and thereby induce the process of repolarisation.
-
NTM : There exist several types of NTMs e.g. small molecules (e.g. acetylcholine, glutamate, GABA), catecholamines (e.g. adrenaline and noradrenaline) and neuropeptides (e.g. neuropeptide Y). After synthesis, these molecules bind transporter molecules that target them to synaptic vesicles for storage and release later on. There exist small and large vesicles for storage : the former (Synaptic Vesicles, SV) contain the classical NTMs (amino acids, dopamine and serotonin) and are released in the synapse; the latter (Large Dense Core Vesicles, LDCV) contain neuropeptides, GF, hormones, amines and can be released at any place along the presynaptic terminals. Release of a NTM load affects the responses of neighbouring neurons to subsequent incoming stimuli (118,265,410).
Current knowledge and obtained results
As Paper 1 illustrates, our group discovered a role for MK5 in F-actin rearrangements in PC12 cells via its in vivo interaction with PKA. F-actin rearrangements are involved in a wide range of processes like motility and differentiation. Our Paper 3 shows differences in exploratory behaviour, which is indicative for anxiousness and mainly involves 5-HT signalling. On the other hand, we observed differences in locomotion, a process that involves dopamine signalling.
An earlier study by Yao et al investigated a role for PKA in vesicle traffic of monoaminergic vesicle transporters (VMAT) in PC12 cells. There exist two types of VMATs which both transport their cargo to LDCVs: VMAT1 mainly transports noradrenaline, whereas VMAT2 transports monoamines (5-HT, dopamine) to the LDCVs. The signal for targeting and localisation to the LDCVs resides in the C-terminal domain of VMAT2. Casein kinase II (CKII) can phosphorylate two serine residues within this signal sequence and this promotes removal from the LDCVs. In PKA-deficient PC12A123.7 cells, where PKA-I and PKA-II show reduced activity compared to the wild-type PC12 cells, VMAT1 targets both small and large vesicles, whereas VMAT2 looses its ability to target LDCVs and mainly delivers its cargo to small vesicles. Transfection of catalytically active PKA restores this missorting (410). This indirect regulation of vesicle traffic leaves an opportunity for other proteins like anchor proteins or kinases like e.g. MK5, to interfere. Especially, since we observe alterations in mouse behaviour which depend on both 5-HT and dopamine signalling, this favours a mechanism at a more basic level.
A microarray study of our inducible PC12 cell line which allows expression of MK5L337A upon doxycycline removal revealed altered gene expression -amongst others- for (unpublished data from Paper 2):
-
K+ voltage gated channels or Kv channels which are involved in the repolarisation after an AP. Some NTM reuptake transporters can interact directly with Kv channels (231). Similarly, the protein interaction map, which we used to assess which proteins could be affected by the changes in gene expression from the microarray, also showed the involvement of these channels and a kinase which regulates KCl co-transporters.
-
Cytochrome p450 regulates retinoic acid (RA) levels. Lower levels of the retinoid receptor seem associated with decreased locomotor activity (204).
-
Chromogranin B is processed in the adrenal medulla from secretogranin I. Chromogranin B contributes to the formation of secretory granules and inhibits parathyroid hormone secretion (PTH). In addition, chromogranin B is reduced in the cerebrospinal fluid of schizophrenic patients (362).
Bibliography
- 1
- N. H.
Acheson.
Fundamentals of Molecular Virology.
John Wiley & Sons Ltd., 2007. - 2
- R. H.
Adams, A. Porras, G. Alonso, M. Jones,
K. Vintersten, S. Panelli, A. Valladares, L. Perez,
R. Klein, and A. R. Nebredda.
Essential role of p38® MAP kinase in placental but not embryonic cardiovascular development.
Molecular Cell, 6:109-116, 2000. - 3
- S. R.
Albright and R. Tjian.
TAFs revisited: more data reveal new twists and confirm old ideas.
Gene, 242(1-2):1-13, 2000. - 4
- D. Allison,
X. Cui, G. Page, and M. Sabripour.
Microarray data analysis: from disarray to consolidation and consensus.
Nature Reviews Genetics, 7(1):55-65, 2006. - 5
- J. R.
Allport and R. Weissleder.
In vivo imaging of gene and cell therapies.
Experimental Hematology, 29:1237-1246, 2001. - 6
- O. Alter,
P. O. Brown, and D. Botstein.
Singular value decomposition for genomewide expression data processing and modeling.
Proc. Natl. Acad. Sci. USA, 97(18):10101-10106, 2000. - 7
- D. R.
Anderson, M. J. Meyers, W. F. Vernier, M. W. Mahoney,
R. G. Kurumbail, N. Caspers, G. I. Poda, J. F.
Schindler, D. B. Reitz, and R. J. Mourey.
Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2).
Journal of Medicinal Chemistry, 50:2647-2654, 2007. - 8
- A. A.
Andrade, P. N. Silva, A. C. Pereira, L. P. DeSousa,
P. C. Ferreira, R. T. Gazzinelli, E. G. Kroon,
C. Ropert, and C. A. Bonjardim.
The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication.
Biochemical Journal, 381:437-446, 2004. - 9
- G. F.
Anhêa, A. S. Torrãoa, T. C. Nogueiraa,
L. C. Caperutoa, M. E. Amaralb, M. C. Medinaa,
A. K. Azevedo-Martinsa, A. R. Carpinellia, C. R.
Carvalhoa, R. Curia, A. C. Boscherob, and S. Bordin.
ERK3 associates with MAP2 and is involved in glucose-induced insulin secretion.
Molecular and Cellular Endocrinology, 251(1-2):33-41, 2006. - 10
- A. F.
Arnsten, B. P. Ramos, S. G. Birnbaum, and J. R. Taylor.
Protein kinase A as a therapeutic target for memory disorders: rationale and challenges.
TRENDS in Molecular Medicine, 11(3):121-128, 2005. - 11
- J. S. C.
Arthur and P. Cohen.
MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells.
FEBS Letters, 482:44-48, 2000. - 12
- J. S. C.
Arthur, A. L. Fong, J. M. Dwyer,
M. Davare, E. Reese, K. Obrietan, and S. Impey.
Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins.
The Journal of Neuroscience, 24(18):4324-4332, 2004. - 13
- J. D.
Ashwell.
The many paths to p38 mitogen-activated protein kinase activation in the immune system.
Nature Reviews Immunology, 6:532-540, 2006. - 14
- E. Asp
and P. Sunnerhagen.
Mkp1 and Mkp2, two MAPKAP-kinase homologues in Schizosaccharomyces pombe, interact with the MAP kinase Sty1.
Mol Genet Genomics, 268:585-597, 2003. - 15
- C. M.
Atkins, J. C. Selcher, J. J. Petraitis, J. M. Trzaskos,
and J. D. Sweatt.
The MAPK cascade is required for mammalian associative learning.
Nature Neuroscience, 1(7):602-609, 1998. - 16
- N. Azuma,
N. Akasaka, H. Kito, M. Ikeda,
V. Gahtan, T. Sasajima, and B. Sumpio.
Role of p38 MAP kinase in endothelial cell alignment induced by fluid shear stress.
Am. J. Physiol. Heart Circ. Physiol, 280:H189-197, 2001. - 17
- A. Baas,
J. Boudeau, G. Sapkota, L. Smit, R. Medema,
N. Morrice, D. Alessi, and H. Clevers.
Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD.
The EMBO Journal, 22(12):3062-3072, 2003. - 18
- A. F.
Baas, J. Kuipers, N. N. van der Wel, E. Batile,
H. K. Koerten, P. J. Peters, and H. C. Clevers.
Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD.
Cell, 116:457-466, 2004. - 19
- H. J.
Baek, Y. K. Kang, and R. G. Roeder.
Human mediator enhances basal transcription by facilitating recruitment of transcription factor IIB during preinitiation complex assembly.
The Journal of Biological Chemistry, 281(22):15172-15181, 2006. - 20
- J. Bain,
H. McLauchlan, M. Elliott, and P. Cohen.
The specificities of protein kinase inhibitors: an update.
Biochemistry Journal, 371:199-204, 2003. - 21
- T. Bakheet,
B. R. Williams, and K. S. Khabar.
ARED 3.0: the large and diverse AU-rich transcriptome.
Nucleic Acids Research, 34:D111-D114, 2006. - 22
- C. Barreau,
L. Paillard, and H. B. Osborne.
AU-rich elements and associated factors: are there unifying principles.
Nucleic Acids Research, 33(22):7138-7150, 2005. - 23
- D. Barsyte-Lovejoy,
A. Galanis, and A. D. Sharrocks.
Specificity determinants in MAPK signaling to transcription factors.
Journal of Biological Chemistry, 277(12):9896-9903, 2002. - 24
- R. Ben-Levy,
S. Hooper, R. Wilson, H. F. Paterson,
and C. J. Marshall.
Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2.
Current Biology, 8:1049-1057, 1998. - 25
- D. Benson,
I. Karsch-Mizrachi, D. Lipman,
J. Ostell, and D. Wheeler.
Genbank.
Nucleic Acids Research, 35(Database issue):D21-5, 2007. - 26
- B. Bethke
and B. Sauer.
Segmental genomic replacement by Cre-mediated recombination: genotoxic stress activation of the p53 promoter in single-copy transformants.
Nucleic Acids Research, 25(14):2828-2834, 1997. - 27
- R. Biddick
and E. T. Young.
Yeast mediator and its role in transcriptional regulation.
C.R. Biologies, 328:773-782, 2005. - 28
- Y. Bilu
and M. Linial.
The advantage of functional prediction based on clustering of Yeast genes and its correlation with non-sequence based classifications.
Journal of Computational Biology, 9(2):193-210, 2002. - 29
- C. bing
Guan, H. tai Xu, M. Jin, X. bing Yuan, and M. ming
Poo.
Long-range Ca2+ signaling from growth cone to soma mediates reversal of neuronal migration induced by Slit-2.
Cell, 129:385-395, 2007. - 30
- C. Birchmeier,
W. Birchmeier, E. Gherardi, and
G. F. V. Woude.
Met, metastasis, motility and more.
Nature Reviews: Molecular Cell Biology, 4:915-925, 2003. - 31
- V. Bissonauth,
S. Roy, M. Gravel, S. Guillemette,
and J. Charron.
Requirement for Map2k1 (Mek1) in extra-embryonic ectoderm during placentogenesis.
Development, 133:3429-3440, 2006. - 32
- C. Bjorbak,
Y. Zhao, and D. E. Moller.
Divergent functional roles for p90RSK kinase domains.
The Journal of Biological Chemistry, 270(32):18848-18852, 1995. - 33
- S. Björklund
and C. M. Gustafsson.
The yeast Mediator complex and its regulation.
TRENDS in Biochemical Sciences, 30(5):240-244, 2005. - 34
- Y. A.
Blednov, M. Stoffel, S. Chang, and R. A. Harris.
Potassium channels as targets for ethanol: Studies of G-protein-coupled inwardly rectifying potassium channel 2 (GIRK2) null mutant mice.
The Journal of Pharmacology and Experimental Therapeutics, 298:521-530, 2001. - 35
- J. Blenis.
Signal transduction via the MAP kinases: Proceed at your own RSK.
Proceedings of the National Academy of Science of the USA, 90:5889-5892, 1993. - 36
- N. Blom,
S. Gammeltoft, and S. Brunak.
Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites.
Journal of Molecular Biology, 294(5):1351-1362, 1999. - 37
- M. A.
Bogoyevitch and N. W. Court.
Counting on mitogen-activated protein kinases - ERKs 3, 4, 5, 6, 7 and 8.
Cellular Signaling, 16:1345-1354, 2004. - 38
- M. A.
Bogue and S. C. Grubb.
The mouse phenome project.
Genetica, 122:71-74, 2004. - 39
- J. Bok,
X. Zha, Y. Cho, and S. Green.
An extranuclear locus of cAMP-dependent protein kinase action is necessary and sufficient for promotion of spiral ganglion neuronal survival by cAMP.
Journal of Neuroscience, 23:777-787, 2003. - 40
- S. Bornstein,
H. Tian, A. Haidan, A. Böttner,
N. Hiroi, G. Eisenhofer, S. McCann, G. Chrousos,
and S. Roffler-Tarlov.
Deletion of tyrosine hydroxylase gene reveals functional interdependence of adrenocortical and chromaffin cell system in vivo.
Proceedings of the National Academy of Science of the USA, 97(26):14724-14747, 2000. - 41
- I. Bossis,
A. Voutetakis, T. Bei, F. Sandrini,
K. Griffin, and C. Stratakis.
Protein kinase A and its rols in human neoplasia: the Carney complex paradigm.
Endocrine-Related Cancer, 11:265-280, 2004. - 42
- T. Boulton,
S. Nye, D. Robbins, N. Ip,
E. Radziejewska, S. Morgenbesser, R. DePinho,
N. Panayotatos, M. Cobb, and G. Yancopoulos.
ERKs: a family of protein ser/thr kinases that are activated and tyrosine phosphorylated in response to insulin and NGF.
Cell, 65(4):663-675, 1991. - 43
- M. Bourin
and M. Hascoët.
The mouse light/dark box test.
European Journal of Pharmacology, 463:55-65, 2003. - 44
- K. Brami-Cherrier,
E. Valjent, D. Hervee,
J. Darragh, J.-C. Corvol, C. Pages, A. J. Simon, J.-A.
Girault, and J. Caboche.
Parsing molecular and behavioral effects of cocaine in Mitogen- and Stress-activated protein Kinase-1-deficient mice.
The Journal of Neuroscience, 25(49):11444-11454, 2005. - 45
- E. P.
Brandon, R. L. Idzerda, and G. S. McKnight.
PKA isoforms, neural pathways, and behaviour: making the connection.
Current Opinion in Neurobiology, 7:397-403, 1997. - 46
- E. P.
Brandon, S. F. Logue, M. A. Adams, M. Qi, S. P.
Sullivan, A. M. Matsumoto, D. M. Dorsa, J. M. Wehner,
G. S. McKnight, and R. L. Idzerda.
Defective motor behavior and neural gene expression in RII¯ -protein kinase A mutant mice.
The Journal of Neuroscience, 18(10):3639-3649, 1998. - 47
- M. Brockington,
D. Blake, S. Brown, and F.Muntoni.
The gene for a novel glycosyltransferase is mutated in congenital muscular dystrophy MDC1C and limb girdle muscular dystrophy 2I.
Neuromuscul Disord, 12(3):233-234, 2002. - 48
- J. P.
Brody, B. A. Williams, B. J. Wod, and S. R. Quake.
Significance and statistical errors in the analysis of DNA microarray data.
Proceedings of the National Academy of Sciences of the USA, 99:12975-12978, 2002. - 49
- W. R.
Burack and A. S. Shaw.
Signal transduction: Hanging on a scaffold.
Current Opinion in Cell Biology, 12:211-216, 2000. - 50
- S. Caenepeel,
G. Charydczak, S. Sudarsanam,
T. Hunter, and G. Manning.
The mouse kinome: Discovery and comparative genomics of all mouse protein kinases.
Proceedings of the National Academy of Sciences of the USA, 101(32):11707-11712, 2004. - 51
- W. Cai,
D.-W. Shin, K. Chen, O. Gheysens, Q. Cao, S. X.
Wang, S. S. Gambhir, and X. Chen.
Peptide-labeled near-infrared quantum dots for imaging tumor vasculature in living subjects.
Nano Letters, 6(4):669-676, 2006. - 52
- J. C.
Carroll, J. M. Boyce-Rustay, R. Millstein, R. Yang,
L. M. Wiedholz, D. L. Murphy, and A. Holmes.
Effects of mild early life stress on abnormal emotion-related behaviors in 5-HTT knockout mice.
Behav. Genet., 37:214-222, 2007. - 53
- E. Casanova,
S. Fehsenfeld, E. Greiner, A. F.
Stewart, and G. Schuetz.
Construction of a conditional allele of RSK-B/MSK2 in the mouse.
Genesis, 32:158-160, 2002. - 54
- S. Chakraborti.
Phospholipase A2 isoforms: a perspective.
Cellular Signaling, 15:637-665, 2003. - 55
- D. Chaturvedi,
H. M. Poppleton, T. Stringfield,
A. Barbier, and T. B. Patel.
Subcellular localization and biological actors of activated RSK1 are determined by its interactions with subunits of cyclic AMP-dependent protein kinase.
Molecular and Cellular Biology, 26(12):4586-4600, 2006. - 56
- A. Chen,
M. Ohno, K. Giese, R. Kuhn, R. Chen, and
A. Silva.
Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory.
Journal of Neuroscience Research, 83:28-38, 2006. - 57
- Y.-Y. Cho,
K. Yao, A. M. Bode, H. R. BergenIII, B. J. Madden,
S.-M. Oh, S. Ermakova, B. S. Kang, H. S. Choi, J.-H.
Shim, and Z. Dong.
RSK2 mediates muscle cell differentiation through regulation of NFAT3.
The Journal of Biological Chemistry, 282(11):8380-8392, 2007. - 58
- S. Choi,
T. Oba, N. Callander, D. Jelinek, and G. Roodman.
AML-1A and AML-1B regulation of MIP-® expression in multiple myeloma.
Blood, 101:3778-3783, 2003. - 59
- C. A.
Chrestensen, M. J. Schroeder, J. Shabanowitz, D. F.
Hunt, J. W. Pelo, M. T. Worthington, and T. W. Sturgill.
MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including ser178, a site required for 14-3-3 binding.
Journal of Biological Chemistry, 279(11):10176-10184, 2004. - 60
- W. S.
Cleveland, E. Grosse, and W. M. Shyu.
Statistical Models in S, chapter Local Regression Models.
Wadsworth & Brooks/Cole, 1992. - 61
- A. D.
Clifton, P. R. Young, and P. Cohen.
A comparison of the substrate specificity of MAPKAPK kinase-2 and MAPKAPK kinase-3 and their activation by cytokines and cellular stress.
FEBS Letters, 392:209-214, 1996. - 62
- S. P.
Collins, J. L. Reoma, D. M. Gamm, and M. D. Uhler.
LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo.
Biochemistry Journal, 345:673-680, 2000. - 63
- R. C.
Conaway, S. Sato, C. Tomomori-Sato, T. Yao, and
J. W. Conaway.
The mammalian Mediator complex and its role in transcriptional regulation.
TRENDS in Biochemical Sciences, 30(5):250-255, 2005. - 64
- M. D.
Conkright, E. Guzman, L. Flechner, A. I. Su, J. B.
Hogenesch, and M. Montminy.
Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness.
Molecular Cell, 11:1101-1108, 2003. - 65
- A. Constantinescu,
M. Wu, O. Asher, and I. Diamond.
cAMP-dependent protein kinase type I regulation ethanol-induced cAMP response element-mediated gene expression via activation of CREB-binding protein and inhibition of MAPK.
The Journal of Biological Chemistry, 279(41):43321-43329, 2004. - 66
- S. Corre
and M. Galibert.
Upstream stimulating factors: highly versatile stress-responsive transcription factors.
Pigment Cell Research, 18:337-343, 2005. - 67
- P. Coulombe,
G. Rodier, S. Pelletier, J. Pellerin,
and S. Meloche.
Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of the mitogen-activated protein kinase regulation during cellular differentiation.
Molecular and Cellular Biology, 23(13):4542-4558, 2003. - 68
- X. Coumoul,
V. Shukla, C. Li, R.-H. Wang, and C.-X.
Deng.
Conditional knockdown of Fgfr2 in mice using Cre-LoxP induced RNA interferences.
Nucleic Acids Research, 33(11):e120, 2005. - 69
- J. N.
Crawley.
What's wrong with my mouse: Behavioral phenotyping of transgenic and knockout mice.
Wiley, second edition, 2007. - 70
- D. Crowe,
R. Kim, and R. Chandraratna.
Retinoic acid differentially regulates cancer cell proliferation via dose-dependent modulation of the mitogen-activated protein kinase pathway.
Molecular Cancer Research, 1:532-540, 2003. - 71
- D. L.
Crowe.
Induction of p97MAPK expression regulates collagen mediated inhibition of proliferation and migration in human squamous cell carcinoma lines.
International Journal of Oncology, 24:1159-1163, 2004. - 72
- B. Cuevas,
A. Abell, and G. Johnson.
Role of mitogen-activated protein kinase kinase kinases in signal integration.
Oncogene, 26:3159-3171, 2007. - 73
- A. Culbert,
S. Skaper, D. Howlett, N. Evans,
L. Facci, P. Soden, Z. Seymour, F. Guillot,
M. Gaestel, and J. Richardson.
MAPKAP kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity: relevance to neuroinflammation in a transgenic mouse model of Alzheimer's disease.
Journal of Biological Chemistry, 281(33):23658-67, 2006. - 74
- D. E.
Cummings, E. P. Brandon, J. V. Planas, K. Motmed,
R. L. Idzerda, and G. S. McKnight.
Genetically lean mice result from targeted dirsuption of the RII subunit of protein kinase A.
subunit of protein kinase A.
Nature, 382:622-626, 1996. - 75
- M. Cyert.
Regulation of nuclear localization during signaling.
The Journal of Biological Chemistry, 276(24):20805-20808, 2001. - 76
- T. Dai,
Y. Vera, E. C. Salido, and P. H. Yen.
Characterization of the mouse DazapI gene encoding an RNA-binding protein that interacts with infertility factors DAZ and DAZL.
BMC Genomics, 2:6, 2001. - 77
- I. Dalle-Donne,
R. Rossi, A. Milzani, P. D.
Simplicio, and R. Colombo.
The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself.
Free Radical Biology and Medicine, 31(12):1624-1632, 2001. - 78
- N. Dard
and M. Peter.
Scaffold proteins in MAP kinase signaling: more than simple passive activating platforms.
Bioessays, 28:146-156, 2006. - 79
- R. Das,
V. Esposito, M. Abu-Abed, G. S. Anand, S. S.
Taylor, and G. Melacini.
cAMP activation of PKA defines an ancient signaling mechanism.
Proceedings of the National Academy of Science of the USA, 104(1):93-98, 2007. - 80
- J. DaSilva,
L. Xu, H. J. Kim, W. T. Miller, and
D. Bar-Sagi.
Regulation of Sprouty stability by Mnk1 dependent phosphorylation.
Molecular and Cellular Biology, 26(5):1898-1907, 2006. - 81
- J.-P. David,
D. Mehic, L. Bakiri, A. F. Schilling, V. Mandic,
M. Priemel, M. H. Idarraga, M. O. Reschke,
O. Hoffmann, M. Amling, and E. F. Wagner.
Essential role of RSK2 in c-Fos-dependent osteosarcoma development.
The Journal of Clinical Investigation, 115(3):664-672, 2005. - 82
- I. Davidson,
D. Kobi, A. Fadloun, and G. Mengus.
New insights into TAFs as regulators of cell cycle and signaling pathways.
Cell Cycle, 4(11):1486-1490, 2005. - 83
- M. Davis.
Role of NMDA receptors and MAP kinase in the amygdala in extinction of fear: clinical implications for exposure therapy.
European Journal of Neuroscience, 16:395-398, 2002. - 84
- R. J.
Davis.
Signal transduction by the JNK group of MAP kinases.
Cell, 103:239-252, 2000. - 85
- R. Day,
J. Walder, and R. Maurer.
A protein kinase inhibitor gene reduces both basal and multihormone-stimulated prolactine gene transcription.
The Journal of Biological Chemistry, 264:431-436, 1989. - 86
- J. G.
de la Nava, S. van Hijum, and O. Trelles.
Saturation and quantization reduction in microarray experiments using two scans at different sensitivities.
Statistical Applications in Genetics and Molecular Biology, 3(1), 2006. - 87
- R. Deacon,
R. Brook, D. Meyer, O.Haeckel,
F. Ashcroft, T. Miki, S. Seino, and B. Liss.
Behavioral phenotyping of mice lacking the KATP channel subunit Kir6.2.
Physiology and Behavior, 87:723-733, 2006. - 88
- M. Deak,
A. D. Clifton, J. M. Lucocq, and D. R. Alessi.
Mitogen-and stress-activated protein kinase-1 (MSK1) is directely activated by MAPK and SAPK2/p38, and may mediate activation of CREB.
The EMBO Journal, 17(15):4426-4441, 1998. - 89
- M. Delghandi,
M. Johannessen, and U. Moens.
The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells.
Cell Signaling, 17:1343-1351, 2005. - 90
- P. Denny,
S. Swift, F. Connor, and A. Ashworth.
An SRY-related gene expressed during spermatogenesis in the mouse encodes a sequence-specific DNA-binding protein.
EMBO Journal, 11:3705-3712, 1992. - 91
- N. DeRuiter,
R. Wolthuis, H. vanDam, B. Burgering,
and J. Bos.
Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor-Ral pathway.
Molecular and Cellular Biology, 20:8480-8488, 2000. - 92
- R. Diskin,
N. Askari, R. Capone, D. Engelberg, and
O. Livnah.
Active mutants of the human p38® MAP kinase.
Journal of Biological Chemistry, 279(45):47040-9, 2004-28/7. - 93
- K. L.
Dodge-Kafka and M. S. Kapiloff.
The mAKAP signaling complex: integration of cAMP, calcium, and MAP kinase signaling pathways.
European Journal of Cell Biology, 85:593-602, 2006. - 94
- D. Drapier,
D. Bentue-Ferrer, B. Laviolle,
B. Millet, H. Allain, M. Bourin, and J. Reymann.
Effects of acute fluoxetine, paroxetine and desipramine on rats tested on the eleveated plus-maze.
Behav Brain Res, 176(2):202-9, 2007. - 95
- S. Dudoit,
Y. H. Yang, M. J. Callow, and T. P.
Speed.
Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments.
Technical report, Department of Biochemistry, Stanford University School, Beckman Center B400, Stanford CA 94305-5307, 2000. - 96
- S. D.
Dufresne, C. Bjørbæk, K. El-Haschimi,
Y. Zhao, W. G. Aschenbach, D. E. Moller, and L. J.
Goodyear.
Altered extrecellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knockout mice.
Molecular and Cellular Biology, 21(1):81-87, 2001. - 97
- N. Dumaz
and R. Marais.
Integrating signals between camp and the ras/raf/mek/erk signalling pathways.
FEBS Journal, 272:3491-504, 2005. - 98
- P. R.
Dunkley, L. Bobrovskaya, M. E. Graham, E. I. von
Nagy-Felsobuki, and P. W. Dickson.
Tyrosine hydroxylase phosphorylation: regulation and consequences.
Journal of Neurochemistry, 91:1025-1043, 2004. - 99
- B. A.
Dümmler, C. Hauge, J. Silbert, H. G. Yntema,
L. S. Kruse, B. Kofoed, B. A. Hemming, D. R.
Alessi, and M. Frödin.
Functional characterization of human RSK4, a new 90-kDa ribosomal S6 kinase, reveals constitutive activation in most cell types.
The Journal of Biological Chemistry, 280(14):13304-13314, 2005. - 100
- D. Eckert.
The AP-2 family of transcription factors.
Genome Biology, 6:246, 2005. - 101
- R. Eferl
and E. Wagner.
AP-1: is a double-edged sword in tumorigenesis.
Nature Reviews Cancer, 3:859-868, 2003. - 102
- H. Einat,
P. Yuan, T. D. Gould, J. Li, J. Du,
L. Zhang, H. K. Manji, and G. Chen.
The role of the extracellular signal-regulated kinase signaling pathway in mood modulation.
The Journal of Neuroscience, 23(19):7311-7316, 2003. - 103
- K. El-Haschimi,
S. D. Dufresne, M. F. Hirshman,
J. S. Flier, L. J. Goodyear, and
C. Bjørbæk.
Insulin resistance and lipodystrophy in mice lacking ribosomal S6 kinase 2.
Diabetes, 52:1340-1346, 2003. - 104
- S. Elbashir,
J. Martinez, A. Patkaniowska,
W. Lendeckel, and T. Tuschl.
Functional anatomy of siRNA for mediating efficient RNAi in Drosophila melanogaster embryo lysate.
The EMBO Journal, 20(23):6877-6888, 2001. - 105
- K. Engel,
A. Kotlyarov, and M. Gaestel.
Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation.
The EMBO Journal, 17(12):3363-3371, 1998. - 106
- D. Evanko.
Bioluminescent quantum dots.
Nature Methods, 3(4):240-241, 2006. - 107
- D. Fambrough,
K. McClure, A. Kazlauskas, and
E. Lander.
Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes.
Cell, 97:727-741, 1999. - 108
- M. H.
Farah.
RNAi silencing in mouse models of neurodegenerative disease.
Current Drug Delivery, 4:161-167, 2007. - 109
- D. Fass,
J. Butler, and R. Goodman.
Deacetylase activity is required for cAMP activation of a subset of CREB target genes.
The Journal of Biological Chemistry, 278:43014-43019, 2003. - 110
- J. E.
FerrellJr.
What do scaffold proteins really do?
Science's STKE, 52:pe1, 2000. - 111
- A. Fischer,
M. Radulovic, C. Schrick, Farahnaz,
Sananbenesi, J. Godovac-Zimmermann, and J. Radulovic.
Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior.
Neurobiology of Learning and Memory, 87(1):149-158, 2007. - 112
- M. Fornerod,
M. Ohno, M. Yoshida, and I. W. Mattaj.
CRM1 is an export receptorf for leucine-rich nuclear export signals.
Cell, 90:1051-1060, 1997. - 113
- J. V.
Frangioni.
Self-illuminating quantum dots light the way.
Nature Biotechnology, 24(3):326-328, 2006. - 114
- L. Friis-Hansen.
Lessons from the gastrin knockout mice.
Regulatory peptides, 139:5-22, 2007. - 115
- S. Fu,
W. Chou, D. Huang, and H. Huang.
Transcriptional regulation of a rice mitogen-activated protein kinase gene, OsMAPK4, in response to environmental stresses.
Plant Cell Physiology, 43:958-963, 2002. - 116
- D. Fusco,
N. Accornero, B. Lavole, S. Shenoy,
J. Blanchard, R. Singer, and E. Bertrand.
Single mRNA molecules demonstrate probabilistic movement in living mammalian cells.
Current Biology, 13:161-1677, 2003. - 117
- M. Gaestel.
MAPKAP kinases - MKs- two's company, three's a crowd.
Nature Reviews Molecular Cell Biology, 7:1-11, 2006. - 118
- W. F.
Ganong.
Review of Medical Physiology.
Appleton and Lange, 19 edition, 1999. - 119
- X. Gao,
Y. Cui, R. M. Levenson, L. W. Chung, and S. Nie.
In vivo cancer targeting and imaging with semiconductor quantum dots.
Nature Biotechnology, 22(8):969-976, 2004. - 120
- N. Gerits,
S. Kostenko, and U. Moens.
In vivo functions of mitogen-activated protein kinases: conclusions from knock-in and knock-out mice.
Transgenic Research, 16:281-314, 2007. - 121
- N. Gerits,
T. Mikalsen, S. Kostenko, A. Shiryaev,
and U. Moens.
Modulation of F-actin rearrangement by the cAMP/PKA pathway is mediated by MAPKAP kinase 5 and requires PKA-induced nuclear export of MK5.
Journal of Biological Chemistry, October 2007.
In Press. - 122
- N. Gerits
and U. Moens.
Protein kinase inhibitors.
Encyclopedia of Molecular Pharmacology, ISBN:978-3-540-38916-3, 2007.
In press. - 123
- N. Gerits,
A. Shiryaev, S. Kostenko, H. Klenow,
O. Shiryaeva, M. Johannessen, and U. Moens.
Mk5 promoter.
2007.
Manuscript. - 124
- D. Gold,
P. Gent, and B. Hamilton.
ROR in genetic control
of cerebellum develoment: 50 staggering years.
Brain research, 1140:19-25, 2007. - 125
- F. A.
Gonzalez, D. L. Raden, M. R. Rigby, and R. J. Davis.
Heterogeneous expression of four MAP kinase isoforms in human tissues.
FEBS, 304(2.3):170-178, 1992. - 126
- Y.-M. Gu,
Y.-H. Jin, J.-K. Choi, K.-H. Baek, C.-Y. Yeo, and K.-Y. Lee.
Protein kinase A phosphorylates and regulates dimerization of 14-3-3 ³.
FEBS Letters, 580:305-310, 2006. - 127
- J. S.
Gutkind.
Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors.
Science's STKE, 40:re1, 2000. - 128
- J. Götz,
N. Deters, A. Doldissen, L. Bokhari,
Y. Ke, A. Ziesner, N. Schonrock, and L. M. Ittner.
A decade of tau transgenic animal models and beyond.
Brain Pathology, 17:91-103, 2007. - 129
- S. Hahn.
Structure and mechanism of the RNA polymerase II transcription machinery.
Nature Structural and Molecular Biology, 11(5):394-403, 2004. - 130
- J. Han
and R. J. Ulevitch.
Emerging targets for anti-inflammatory therapy.
Nature Cell Biology, 1:E39-40, 1999. - 131
- L. Han,
D. Wong, G. Tsai, Z. Jiang, and J. Coyle.
Promoter analysis of human glutamate carboxypeptidase II.
Brain Research, 1170:1-12, 2007. - 132
- S. K.
Hanks.
Genomic analysis of the eukaryotic protein kinase superfamily.
Genome Biology, 4(5):111, 2003. - 133
- M. Hannigan,
L. Zhan, Y. Ai, A. Kotlyarov,
M. Gaestel, and C. Huang.
Abnormal migration phenotype of mitogen-activated protein kinase-activated protein kinase 2 neutrophils in Zigmond chambers containing
formyl-methionyl-leucyl-phenylalanine gradients.
neutrophils in Zigmond chambers containing
formyl-methionyl-leucyl-phenylalanine gradients.
Journal of Immunology, 167:3953-3961, 2001. - 134
- N. Hatano,
Y. Mori, M. Oh-hora, A. Kosugi,
T. Fujikawa, N. Nakai, H. Niwa, J. ichi Miyazaki,
T. Hamaoka, and M. Ogata.
Essential role for ERK2 mitogen-activated protein kinase in placental development.
Genes to Cells, 8:847-856, 2003. - 135
- J. C.
Hedges, M. A. Dechert, I. A. Yamboliev, J. L. Martin,
E. Hickey, L. A. Weber, and W. T. Gerthoffer.
A role for p38MAPK/HSP27 pathway in smooth muscle cell migration.
The Journal of Biological Chemistry, 274(34):24211-24219, 1999. - 136
- D. Heffron
and J. W. Mandell.
Differential localization of MAPK-activated protein kinases RSK1 and MSK1 in mouse brain.
Molecular Brain Research, 136:134-141, 2005. - 137
- Y. Hefner,
A. G. Boersch-Haubold, M. Murakami,
J. I. Wilde, S. Pasquet, D. Schieltz,
F. Ghomashechi, J. R. YatesIII, C. G. Armstrong,
A. Paterson, P. Cohen, R. Fukunaga, T. Hunter,
I. Kudo, S. P. Watson, and M. H. Gelb.
Serine 727 phosphorylation and activation of phospholipase A2 by MNK1-related protein kinase.
The Journal of Biological Chemistry, 275(48):37542-37551, 2000. - 138
- M. Hegen,
M. Gaestel, C. Nickerson-Nutter, L. Lin,
and J. Telliez.
MAPKAP kinase 2-deficient mice are restistant to collagen-induced arthritis.
The Journal of Immunology, 177(3):1913-1917, 2006. - 139
- N. Heintzman
and B. Ren.
The gateway to transcription: identifying, characterizing and understanding promoters in the eukaryotic genome.
Cellular and Molecular Life Sciences, 64:386-400, 2007. - 140
- E. J.
Helmreich.
Biochemistry of Cell Signaling.
Oxford Press, 2001. - 141
- H. Hermeking
and A. Benzinger.
14-3-3 proteins in cell cycle regulation.
Seminars in Cancer Biology, 16:183-192, 2006. - 142
- C. Hetzer,
D. Bisgrove, M. S. Cohen, A. Pedal,
K. Kaehlcke, A. Speyerer, K. Bartscherer,
J. Taunton, and M. Ott.
Recruitment and activation of RSK2 by HIV-1 Tat.
PLoS ONE, 2(1):e51, 2007. - 143
- R. C.
Hillig, U. Eberspaecher, F. Monteclaro, M. Huber,
D. Nguyen, A. Mengel, B. Muller-Tiemann, and
U. Egner.
Structural basis for a high affinity inhibitor bound to protein kinase MK2.
Journal of Molecular Biology, 369:735-745, 2007. - 144
- T. Hirabayashi,
T. Myrayama, and T. Shimizu.
Regulatory mechanism and physiological role of cytosolic phospholipase A2.
Biological and Pharmaceutical Bulletin, 27(8):1168-1173, 2004. - 145
- E. Hitti,
T. Iakovleva, M. Brook, S. Deppenmeier,
A. D. Gruber, D. Radzioch, A. R. Clark, P. J.
Blackshear, A. Kotlyarov, and M. Gaestel.
Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraproline expression, stability and binding to adenine/uridine-rich element.
Molecular and Cellular Biology, 26(6):2399-2407, 2006. - 146
- A. Holmes,
Q. Li, D. Murphy, E. Gold, and
J. Crawley.
Abnormal anxiety-related behavior in serotonin transporter null mutant mice: the influence of genetic background.
Genes, Brain and Behavior, 2:365-380, 2003. - 147
- A. Holmes
and R. Rodgers.
Influence of spatial and temporal manipulations on the anxiolytic efficacy of chlordiazepoxide in mice previously exposed to the elevated plus-maze.
Neuroscience and Biobehavioral Reviews, 23:971-980, 1999. - 148
- J. D.
Hommel, R. M. Sears, D. Georgescu, D. L. Simmons, and
R. J. DiLeone.
Local gene knockdown in the brain using viral-mediated RNA interference.
Nature Medicine, 9(12):1539-1544, 2003. - 149
- W. Hong,
C. He, L. Wang, D. Wang, L. Joseph,
C. Jantasuriyarat, L. Dai, and G. Wang.
BWMK1 responds to multiple environmental stresses and plant hormones.
Journal of Integrative Plant Biology, 49:843-851, 2007. - 150
- R. Houben,
J. Becker, and U. Rapp.
Absence of mutations in the coding sequence of the potential tumor suppressor 3pK in metastatic melanoma.
Journal of Carcinogenesis, 20:23, 2005. - 151
- M. D.
Houslay.
A RSK(y) relationship with promiscuous PKA.
Science's STKE, 349:pe32, 2006. - 152
- A. K.
Howe.
Regulation of actin-based cell migration by cAMP/PKA.
Biochimica et Biophysica Acta, 1692:159-174, 2004. - 153
- C. Huang,
K. Jacobson, and M. Schaller.
MAP kinases and cell migration.
Journal of Cell Science, 117:4619-4628, 2004. - 154
- H. Huang,
S. Fu, Y. Tai, W. Choi, and
D. Huang.
Expression of Oryza sativa MAP kinase gene is developmentally regulated and stress-responsive.
Physiologica Plantarum, 114:572-580, 2002. - 155
- Y. Huang,
H. Roelink, and G. S. McKnight.
Protein kinase A deficiency causes axially localized neural tube defects in mice.
The Journal of Biological Chemistry, 277(22):19889-19896, 2002. - 156
- W. Huber,
A. von Heydebreck, and M. Vingron.
Error models for microarray intensities.
In M. Dunn, L. Jorde, P. Little, and S. Subramaniam, editors, Encyclopedia of Genomics, Proteomics and Bioinformatics. John Wiley & Sons Ltd., March 2004. - 157
- J. P.
Hughes, P. C. Staton, M. G. Wilkinson, P. J. L. M.
Strijbos, S. D. Skaper, J. S. C. Arthur, and A. D.
Reith.
Mitogen and stress response kinase-1 (MSK1) mediates excitotoxic induced death of hippocampal neurones.
Journal of Neurochemistry, 86:25-32, 2003. - 158
- J. Huot,
F. Houle, F. Marceau, and J. Landry.
Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells.
Circulation Research, 80:383-392, 1997. - 159
- J. Huot,
F. Houle, S. Rousseau, R. G. Deschesnes, G. M.
Shah, and J. Landry.
SAPK2/p38-dependent F-actin reorganization regulates early membrane blebbing during stress-induced apoptosis.
The Journal of Cell Biology, 143(5):1361-1373, 1998. - 160
- C. Iavarone,
M. Acunzo, F. Carlomagno, A. Catania,
R. M. Melillo, S. M. Carlogmagno, M. Santoro, and
M. Chiariello.
Activation of the Erk8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutive active form of the RET proto-oncogene.
The Journal of Biological Chemistry, 281(15):10567-10576, 2006. - 161
- K. ichi
Jishage, J. ichi Nezu, Y. Kawase, T. Iwata,
M. Watanabe, A. Miyoshi, A. Ose, K. Habu,
T. Kake, Nobuo, Kamada, O. Ueda, M. Kinoshita,
D. E. Jenne, M. Shimane, and H. Suzuki.
Role of Lkb1, the causative gene of Peutz-Jegher's syndrome, in embryogenesis and polyposis.
Proceedings of the National Academy of Sciences of the USA, 99(13):8903-8908, 2002. - 162
- T. Ideker,
V. Thorsson, A.F.Siegel, and L.E.Hood.
Testing for differentially expressed genes by maximum-likelihood analysis of microarray data.
Journal of Computational Biology, 7:805-818, 2000. - 163
- L. M.
Igaz, M. R. M. Vianna, J. H. Medina, and
I. Izquierdo.
Two time periods of hippocampal mRNA synthesis are required for memory consolidation of fear-motivated learning.
The Journal of Neuroscience, 22(15):6781-6789, 2002. - 164
- J. N.
Ihle and H. Hughes.
The challenges of the translating knockout phenotypes into gene function.
Cell, 102:131-134, 2000. - 165
- M. Imagawa,
R. Chiu, and M. Karin.
Transcription factor AP-2 mediates induction by two different signal-transduction pathways: protein kinase C and cAMP.
Cell, 51:251-260, 1987. - 166
- S. Imagawa,
S. Fujii, J. Dong, T. Furumoto,
T. Kaneko, T. Z. Y. Satoh, Tsutsui, and B. Sobel.
Hepatocyte growth factor regulated E box-dependent plasminogen activator inhibitor type 1 gene expression in HepG2 liver cells.
Arteriosclerosis, Thrombosis and Vascular Biology, 26:2407-2413, 2006. - 167
- M. Imajo,
Y. Tsuchiya, and E. Nishida.
Regulatory mechanisms and functions of MAP kinase signaling pathways.
IUBMB Life, 58(5-6):312-317, 2006. - 168
- S. Impey,
S. R. McCorkle, H. Cha-Molstad, J. M.
Dwyer, G. S. Yochum, J. M. Boss, S. McWeeney, J. J.
Dunn, G. Mandel, and R. H. Goodman.
Defining the CREB regulon: a genome-wide analysis of transcription factory regulatory regions.
Cell, 119:1041-1054, 2004. - 169
- W. F.
Inc.
Wikipedia: the free encyclopedia.
http://www.wikipedia.org, 2001-2007. - 170
- D. Inoue,
M. Ohe, Y. Kanemori, T. Nobui, and
N. Sagata.
A direct link of the Mos-MAPK pathway to Erp1/Emi2 in meiotic arrest of Xenopus laevis eggs.
Nature, 446:1100-1104, 2007. - 171
- M. Ishida,
T. Ishida, H. Nakashima, N. Miho,
K. Miyagawa, K. Chayama, T. Oshima, M. Kambe, and
M. Yoshizumi.
Mnk1 is required for angiotensin II-induced protein synthesis in vascular smooth muscle cells.
Circulation Research, 93:1218-1224, 2003. - 172
- T. Iwakuma
and G. Lozano.
Crippling p53 activities via knock-in mutations in mouse models.
Oncogene, 26:2177-2184, 2007. - 173
- G. Izidio,
L. SpricigoJr, and A. Ramos.
Genetic differences in the elevated plus-maze persist after first exposure of inbred rats to the test apparatus.
Behavioural Processes, 68:129-134, 2005. - 174
- R. Janknecht.
Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1).
Oncogene, 22:746-755, 2003. - 175
- R. Jauch,
S. Jäkel, C. Netter, K. Schreiter,
B. Aicher, H. Jäckle, and M. C. Wahl.
Crystal structures of the Mnk2 kinase domain reveal an inhibitory conformation and a zinc binding site.
Structure, 13:1559-1568, 2005. - 176
- H. Ji,
M. R. Ramsey, D. N. Hayes, C. Fan, K. McNamara,
P. Kozlowski, C. Torrice, M. C. Wu, T. Shimamura,
S. A. Perera, M.-C. Liang, D. Cai, G. N. Naumov,
L. Bao, C. M. Contreras, D. Li, L. Chen,
J. Krishnamurthy, J. Koivunen, L. R. Chirieac,
R. F. Padera, R. T. Bronson, N. I. Lindeman, D. C.
Christiani, X. Lin, G. I. Shapiro, P. A. Jänne,
B. E. Johnson, M. Meyerson, D. J. Kwiatkowski,
D. H. Castrillon, N. Bardeesy, N. E. Sharpless, and
K.-K. Wong.
LKB1 modulates lung cancer differentiation and metastasis.
Nature Letters, 448:807-811, 2007. - 177
- M. Johannessen,
M. Delghandi, A. Rykx, A. M.
Dragset, J. Vandenheede, J. V. Lint, and U. Moens.
Protein kinase D induces transcription through direct phosphorylation of the cAMP response element-binding protein.
Journal of Biological Chemistry, 282:14777-14787, 2007. - 178
- M. Johannessen,
M. P. Delghandi, and U. Moens.
What turns CREB on?
Cellular Signalling, 16:1211-1227, 2004. - 179
- M. Johannessen,
M. P. Delghandi, O. M. Seternes,
B. Johansen, and U. Moens.
Synergistic activation of CREB-mediated transcription by forskolin and phorbol ester requires PKC and depends on the glutamine-rich Q2 transactivation domain.
Cellular Signalling, 16:1187-1199, 2004. - 180
- M. Johannessen
and U. Moens.
Trends in Cellular Signalling, chapter Transcription of genes in response to activated cAMP/protein kinase A signalling pathway : There is more to it than CREB.
Nova Publisher, 2005. - 181
- M. Johannessen
and U. Moens.
Multisite phosphorylation of the cAMP response element-binding protein (CREB) by a diversity of protein kinases.
Frontiers in Biosciences, 12:1814-1832, 2007. - 182
- C. Johansen,
A. T. Funding, K. Otkjær,
K. Kragballe, U. B. Jensen, M. Madsen, L. Binderup,
T. Skak-Nielsen, M. S. Fjording, and L. Iversen.
Protein expression of TNF-® in psoriatic skin is regulated at a posttranscriptional level by MAPK-activated protein kinase 2.
The Journal of Immunology, 176:1431-1438, 2006. - 183
- R. Juliano,
P. Reddig, S. Alahari, M.Edin, A. Howe,
and A. Aplin.
Integrin regulation of cell signalling and motility.
Molecular Environment of Integrins, 32(3):443-446, 2004. - 184
- C. Julien,
P. Coulombe, and S. Meloche.
Nuclear export of ERK3 by a CRM1-dependent mechanism regulates its inhibitory action on cell cycle progression.
The Journal of Biological Chemistry, 278(43):42615-42624, 2003. - 185
- G. M.
Kammer.
Mechanisms of deficient type I protein kinase A activity in Lupus T lymphocytes.
International Reviews of Immunology, 23:225-244, 2004. - 186
- P. Kannan-Thulasiraman,
A. Katsoulidis, M. S. Tallman,
J. S. C. Arthur, and L. C. Platanias.
Activation of the mitogen- and stress-activated kinase 1 by aresenic trioxide.
The Journal of Biological Chemistry, 281(32):22446-22462, 2006. - 187
- S. Kant,
S. Schumacher, M. K. Singh, A. Kispert,
A. Kotlyarov, and M. Gaestel.
Characterization of the atypical MAP kinase ERK4 and its activation of the MAPK-activated protein kinase MK5.
Journal of Biological Chemistry, 281(46):35511-9, 2006. - 188
- P. Karuman,
O. Gozani, R. D. Odze, X. C. Zhou,
H. Zhu, R. Shaw, T. P. Brien, C. D. Bozzuto,
D. Ooi, L. C. Cantley, and J. Yuan.
The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death.
Molecular Cell, 7:1307-1319, 2001. - 189
- U. Kayyali,
C. Pennella, C. Trujillo, O.Villa,
M. Gaestel, and P. Hassoun.
Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2.
Journal of Biological Chemistry, 277:42596-42602, 2002. - 190
- P. Killeen.
An alternative to null-hypothesis significance tests.
Psychological science, 16(5):345-53, 2005. - 191
- C. Kim,
D. Vigil, G. Anand, and S. S. Taylor.
Structure and dynamics of PKA signaling proteins.
European Journal of Cell Biology, 85:651-654, 2006. - 192
- H. S.
Kim, S. Yumkham, S.-H. Kim, K. Yea, Y. C. Shin,
S. H. Ryu, and P.-G. Suh.
Secretin induces neurite outgrowth of PC12 through cAMP-mitogen-activated protein kinase pathway.
Experimental and Molecular Medicine, 38(1):85-93, 2006. - 193
- S. Kim,
J. Hong, S. Ryu, W. Chung, J. Yoon, and
J. Koo.
Regulation of mucin gene expression by CREB via a nonclassical retinoic acid signalling pathway.
Molecular and Cellular Biology, 27:6933-6947, 2007. - 194
- A. Kistner,
M. Gossen, F. Zimmerman, J. Jerecic,
C. Ullmer, H. Lübbert, and H. Bujard.
Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice.
Proceedings of the National Academy of Sciences of the USA, 93:10933-10938, 1996. - 195
- K. Kitta,
R. Day, Y. Kim, I. Torregroza,
T. Evans, and Y. Suzuki.
Hepatocyte growth factor induces GATA-4 phosphorylation and cell survival in cardiac muscle cells.
The Journal of Biological Chemistry, 278:4705-4712, 2002. - 196
- A. Klink,
K. Schiebel, M. Winkelmann, E. Rao,
B. Horsthemke, H.-J. Lüdecke, U. Claussen,
G. Scherer, and G. Rappold.
The human protein kinase gene PKX1 on Xp22.3 displays Xp/Yp homology and is a site of chromosomal instability.
Human Molecular Genetics, 4(5):869-878, 1995. - 197
- W. S.
Klung, M. R. Cummings, and C. A. Spencer.
Concepts of Genetics.
Pearson/Prentice Hall, 8 edition, 2006. - 198
- M. Kobayashi,
M. Nishita, T. Mishima, and K. O. adn
Kensaku Mizuno.
MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration.
The EMBO Journal, 25:713-726, 2006. - 199
- M. Kohn,
H. Hameister, M. Vogel, and H. Kehrer-Sawatzki.
Expression pattern of the Rsk2, Rsk4 and Pdk1 genes during murine embryogenesis.
Gene Expression Patterns, 3:173-177, 2003. - 200
- S. Komatsu,
N. Murai, G. Totsukawa, M. Abe,
K. Akasaka, H. Shimada, and H. Hosoya.
Identification of MAPKAPK homolog (MAPKAPK-4) as a myosin II regulatory light-chain kinase in sea urchin egg extracts.
Archives of Biochemistry and Biophysics, 343(1):55-62, 1997. - 201
- A. Kotlyarov,
A. Neininger, C. Schubert, R. Eckert,
C. Birchmeier, H.-D. Volk, and M. Gaestel.
MAPKAP kinase 2 is essential for LPS-induced TNF-® biosynthesis.
Nature Cell Biology, 1:94-97, 1999. - 202
- A. Kotlyarov,
Y. Yannoni, S. Fritz, K. Laa¯
, J.-B. Telliez, D. Pitman,
L. ling Lin, and M. Gaestel.
Distinct cellular functions of MK2.
Molecular and cellular Biology, 22(13):4827-4835, 2002. - 203
- T. Kouzarides.
Acetylation: a regulatory modification to rival phosphorylation?
The EMBO Journal, 19(6):1176-1179, 2000. - 204
- W. Krezel,
N. Ghyselinck, T. A. Samad, V. Dupe,
P. Kastner, E. Borrelli, and P. Chambon.
Impaired locomotion and dopamine signaling in retinoid receptor mutant mice.
Science, 279:863-867, 1998. - 205
- M. Kubista.
Experimental correction for the inner-filter effect in fluorescence spectra.
Analyst, 119:417-419, 1994. - 206
- E. J.
Kuhn and P. K. Geyer.
Genomic insulators: connecting properties to mechanism.
Current Opinion in Cell Biology, 15:259-265, 2003. - 207
- T. Kurosu,
A. I. Hernandez, and J. H. Schwartz.
Serotonin induces selective cleavage of the PKA RI subunit but not RII subunit in Aplysia neurons.
Biochemical and Biophysical Research Communications, 2007. - 208
- M. Kurt,
A. Arik, and S. Celik.
The effects of sertraline and fluoxetine on anxiety in the elevated plus-maze test in mice.
J Basic Clin Physiol Pharmacol, 11(2):173-80, 2000. - 209
- J. M.
Kyriakis and J. Avruch.
Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation.
Physiological Reviews, 81(2):807-867, 2001. - 210
- L. S.
Lambeth, R. J. Moore, M. Muralitharan, B. P. Dalrymple,
S. McWilliams, and T. J. Doran.
Characterisation and application of a bovine U6 promoter for expression of short hairpin RNAs.
BMC Biotechnology, 5:13, 2005. - 211
- J. Larsen,
I. Yamboliev, L. Weber, and
W. Gerthoffer.
Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle.
American Journal of Physiology, 273:L930-L940, 1997. - 212
- W. Lee,
P. Mitchell, and R. Tjian.
Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements.
Cell, 49:741-752, 1987. - 213
- M. D.
Lehner, F. Schwoebel, A. Kotlyarov, M. Leist,
M. Gaestel, and T. Hartung.
Mitogen-activated protein kinase-activated protein kinase 2-deficient mice show increased susceptibility to Listeria monocytogenes infection.
Journal of Immunology, 168:4667-4673, 2002. - 214
- M. Lemaire,
C. Froment, R. Boutros, O. Mondesert,
A. R. Nebreda, B. Monsarrat, and B. Ducommun.
Cdc25B phosphorylation by p38 and MK-2.
Cell cycle, 5(15):1649-1653, 2006. - 215
- K. Lesch
and R. Mössner.
Inactivation of 5HT transport in mice: Modeling altered 5HT homeostasis implicated in emotional dysfunction, affective disorders and somatic syndromes.
Handbook of Experimental Pharmacology, 175:417-456, 2006. - 216
- B. Lewin.
Genes VII.
Oxford University Press, 7 edition, 2000. - 217
- G. Li,
J. J. Lucas, and E. W. Gelfand.
Protein kinase C alpha, betaI and betaII isozymes regulate cytokine production in mast cells through MEKK2/ERK5-dependent and -independent pathway.
Cellular Immunology, 238(1):10-8, 2005. - 218
- Y. Li,
K. Inoki, P. Vacratsis, and K.-L. Guan.
The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product and enhances its interaction with 14-3-3.
The Journal of Biological Chemistry, 278(16):13663-13671, 2003. - 219
- A. Lieber
and M. A. Kay.
Adenovirus-mediated expression of ribozymes in mice.
Journal of Virology, 70(5):3153-3158, 1996. - 220
- D. Livak
and T. Schmittgen.
Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method.
Methods, 25(4):402-408, 2001. - 221
- K. Lo,
N. Landau, and S. Smale.
LyF-1, a transcriptional regulator that interacts with a novel class of promoters for lymphocyte-specific genes.
Molecular and Cellular Biology, 11:5229-5243, 1991. - 222
- H. Lodish,
D. Baltimore, A. Berk, S. L. Zipursky,
P. Patsudaira, and J. Darnell.
Molecular Cell Biology.
Scientific American Books, third edition, 1995. - 223
- A. O.
Loghlen, V. M. Gonzalez, T. Jurado, M. Salinas, and
M. E. Martin.
Characterization of the activity of human MAP kinase-interacting kinase Mnk1b.
Biochimica et Biophysica Acta, 1773(9):1416-27, 2007. - 224
- J. Lowry
and J. Mackay.
GATA-1: one protein, many partners.
Internation Journal of Biochemistry and Cell Biology, 38:6-11, 2006. - 225
- T. Luft,
G. S. Pereira, M. Cammarota, and I. Izquirdo.
Different time course for the memory facilitating effect of bicuculline in hippocampus, entorhinal cortex and posterior parietal cortex of rats.
Neurobiology of Learning and Memory, 82:52-56, 2004. - 226
- B. Luscher,
P. Mitchell, T. Williams, and R. Tjian.
Regulation of transcription factor AP-2 by the morphogen retinoic acid and by second messengers.
Genes and Development, 3:1507-1517, 1989. - 227
- R. J.
Lye and B. T. Hinton.
Transgenic technologies for the study of epididymal function.
Asian Journal of Andrology, 2:33-38, 2000. - 228
- H. Lyng,
A. Badiee, D. H. Svendsrud, E. Hovig, O. Myklebost,
and T. Stokke.
Profound influence of microarray scanner characteristics on gene expression ratios: analysis and procedure for correction.
BMC Genomics, 5(10), 2004. - 229
- S. Ma
and H. Bohnert.
Integration of Arabidopsis thaliana stress-related transcript profiles, promoter structures, and cell-specific expression.
Genome Biology, 8:R49, 2007. - 230
- S. Ma,
G. Liu, Y. Sun, and J. Xie.
Relocalization of the polypyrimidine tract-binding protein during pka-induced neurite growth.
Biochimica et Biophysica Acta, 1773(6):912-923, 2007. - 231
- R. Maiya,
I. Ponomarev, K. Linse, R. Harris, and
R. Mayfield.
Defining the dopamine transporter proteome by convergent biochemical and in silico analyses.
Genes, Brain and Behavior, 6:97-106, 2007. - 232
- E. T.
Maizels, A. Mukherjee, G. Sithanandam, C. A. Peters,
J. Cottom, K. E. Mayo, and M. Hunzicker-Dunn.
Developmental regulation of mitogen-activated protein kinase-activated kinases-2 and -3 (MAPKAPK-2/-3) in vivo during corpus luteum formation in the rat.
Molecular Endocrinology, 15(5):716-733, 2001. - 233
- I. A.
Manke, A. Nguyen, D. Lim, M. Q. Stewart, A. E.
Elia, and M. B. Yaffe.
MAPKAPK2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation.
Molecular Cell, 17:37-48, 2005. - 234
- G. Manning,
D. Whyte, R. Martinez, T. Hunter, and
S. Sudarsanam.
The protein kinase complement of the human genome.
Science, 298:1912-1934, 2002. - 235
- M. Mayford,
M. E. Bach, Y.-Y. Huang, L. Wang, R. D.
Hawkins, and E. R. Kandel.
Control of memory formation through regulated expression of a CaMKII transgene.
Science, 274(5293):1678-1683, 1996. - 236
- A. M.
Mazarati, J. G. Hohmann, A. Bacon, H. Liu,
R. Sankar, R. A. Steiner, D. Wynick, and C. G.
Wasterlain.
Modulation of hippocampal excitability and seizures by galanin.
The Journal of Neuroscience, 20(16):6276-6281, 2000. - 237
- C. Mazzucchelli,
C. Vantaggiato, A. Ciamei,
S. Fasano, P. Pakhotin, W. Krezel, H. Welzl,
D. P. Wolfer, G. Pagès, O. Valverde,
A. Marowsky, A. Porrazzo, P. C. Orban,
R. Maldonado, M. U. Ehrengruber, V. Cestari, H.-P. Lipp,
P. F. Chapman, J. Pouysségur, and R. Brambilla.
Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory.
Neuron, 34:807-820, 2002. - 238
- C. E.
McCoy, D. G. Campbell, M. Deak, G. B. Bloomberg, and
J. S. C. Arthur.
MSK1 activity is controlled by multiple phosphorylation sites.
Biochemistry Journal, 387:507-517, 2005. - 239
- M. McMahon.
Neural patterning: the role of Nkx genes in the ventral spinal cord.
Genes and Development, 14:2261-2264, 2000. - 240
- W. Meng,
L. L. Swenson, M. J. Fitzgibbon, K. Hayakawa,
E. ter Haar, A. E. Behrens, J. R. Fulghum, and
J. A. Lippke.
Structure of mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 suggests a bifunctional switch that couples kinase activation with nuclear export.
The Journal of Biological Chemistry, 277(40):37401-37405, 2002. - 241
- K. Merienne,
S. Jacquot, M. Zeniou, S. Pannetier,
P. Sassone-Corsi, and A. Hanauer.
Activation of RSK by UV-light: phosphorylation dynamics and involvement of the MAPK pathway.
Oncogene, 19:4221-4229, 2000. - 242
- K. Merienne,
S. Pannetier, A. Harel-Bellan, and
P. Sassone-Corsi.
Mitogen-regulated RSK2-CBP interaction controls their kinase and acetylase activities.
Molecular and Cellular Biology, 21(20):7089-7096, 2001. - 243
- T. Mikalsen,
N. Gerits, and U. Moens.
Inhibitors of signal transduction protein kinases as targets for cancer therapy.
Biotechnology Annual Review, 16:153-223, 2006. - 244
- T. Mikalsen,
M. Johannessen, and U. Moens.
Sequence- and position-dependent tagging protects extracellular-regulated kinase 3 protein from 26S proteasome-mediated degradation.
International Journal of Biochemistry and Cell Biology, 37:2513-2520, 2005. - 245
- A. H.
Milton, N. Khaire, L. Ingram, A. J. O'Donnell, and
N. B. LaThangue.
14-3-3 proteins integrate E2F activity with the DNA damage response.
The EMBO Journal, 25:1046-1057, 2006. - 246
- Y. Mizoguchi,
K. Irie, T. Hirayama, N. Hayashida,
K. Yamaguchi-Shinozaki, K. Matsumoto, and K. Shinozaki.
A gene encoding a mitogen-activated protein kinase kinase kinase is induced simultaneously with genes for a mitogen-activated protein kinase and an S6 ribosomal protein kinase by touch, cold, and water stress in Arabidopsis thaliana.
Proceedings of the National Academy of Sciences of the USA, 93:765-769, 1996. - 247
- U. Moens,
N. Subramaniam, B. Johansen, T. Johansen,
and T. Traavik.
A steroid hormone response unit in the late leader of the noncoding control region of the human polyomavirus bk confers enhanced host cell permissivity.
Journal of Virology, 68:2398-2408, 1994. - 248
- M. Montminy.
Transcriptional regulation by cyclic AMP.
Annual Reviews in Biochemistry, 66:807-822, 1997. - 249
- K. Morano
and D. Thiele.
Heat shock factor function and regulation in response to cellular stress, growth, and differentiation signals.
Gene Expression, 7:271-282, 1999. - 250
- D. K.
Morrison and R. J. Davis.
Regulation of MAPK modules by scaffold proteins in mammals.
Annual Reviews in Cellular and Developmental Biology, 19:91-118, 2003. - 251
- S. Morton,
H.-T. Yang, N. Moleleki, D. G. Campbell,
P. Cohen, and S. Rousseau.
Phosphorylation of the ARE-binding protein DAZAP1 by ERK2 induces its dissociation from DAZ.
Biochemical Journal, 399:265-273, 2006. - 252
- C. I.
Mourin and J. Huot.
Recent advances in stress signaling in cancer.
Cancer Research, 64:1893-1878, 2004. - 253
- J. S.
Mudgett, J. Ding, L. Guh-Siesel, N. A. Chartrain,
L. Yang, S. Gopal, and M. M. Chen.
Essential role for p38
mitogen-activated protein kinase in placental angiogenesis.
Proceedings of the National Academy of Sciences, 97:10454-10459, 2000. - 254
- R. Mujumdar,
L. Ernst, S. Mujumdar, C. Lewis, and
A. Waggoner.
Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters.
Bioconjug Chem, 4(2):105-111, Mar-Apr 1993. - 255
- K. S.
Nadella and L. S. Kirschner.
Disruption of protein kinase A regulation causes immortalization and dysregulation of D-type cyclins.
Cancer Research, 65(22):10307-15, 2005. - 256
- A. Nakaya,
S. Goto, and M. Kanehisa.
Extraction of correlated gene clusteres by multiple graph comparison.
Genome Inform., 12:44-53, 2001. - 257
- D. Natale,
A. Paliga, F. Beier, S. D'Souza, and
A. Watson.
p38 MAPK signaling during murine preimplantation development.
Developmental Biology, 268(1):76-88, 2004. - 258
- A. Naär,
B. L. BD, and R. Tjian.
Transcriptional coactivator complexes.
Annual Reviews in Biochemistry, 70:475-501, 2001. - 259
- M. V.
Nesterova and Y. S. Cho-Chung.
Chemoprevention with protein kinase A RI® antisense in DMBA-mammary carcinogenesis.
Annals of the New York Academy of Science, 1058:255-264, 2005. - 260
- B. Neufeld,
A. Grosse-Wilde, A. Hoffmeyer, B. W.
Jordan, P. Chen, D. Dinev, S. Ludwig, and U. R.
Rapp.
Serine/threonine kinases 3pK and MAPK-acitvated protein kinase 2 interact with basic helix-loop-helix transcription factor E47 and repress its transcriptional activity.
The Journal of biological Chemistry, 275(27):20239-20242, 2000. - 261
- L. New,
Y. Jiang, and J. Han.
Regulation of PRAK subcellular location by p38 MAPK.
Molecular Biology of the Cell, 14:2603-2616, 2003. - 262
- L. New,
Y. Jiang, M. Zhao, K. Liu, W. Zhu, L. J.
Flood, Y. Kato, G. C. Parry, and J. Han.
PRAK, a novel protein kinase regulated by p38 MAPK.
The EMBO Journal, 17(12):3372-3384, 1998. - 263
- A. C.
Newton.
Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm.
Biochemical Journal, 307:361-371, 2003. - 264
- H. Ni,
X. Wang, K. Diener, and Z. Yao.
MAPKAPK5, a novel MAPK activated protein kinase is substrate of the extracellular regulated kinase (ERK) and p38 kinase.
Biochemical and Biophysical Research Communications, 243:492-496, 1998. - 265
- D. G.
Nicholls.
Proteins, transmitters and synapses.
Blackwell Science, 1994. - 266
- K. Niefind
and O.-G. Issinger.
Primary and secondary interactions between CK2® and CK2¯ to ring-like structures in the crystals of the CK2 holoenzyme.
Molecular and Cellular Biochemistry, 274:3-14, 2005. - 267
- D. No,
T.-P. Yao, and R. M. Evans.
Ecdysone-inducible gene expression in mammalian cells and transgenic mice.
Proceedings of the National Academy of Sciences of the USA, 93:3346-3351, 1996. - 268
- M. A.
Nolan, D. F. Babcock, G. Wennemuth, W. Brown, K. A.
Burton, and G. S. McKnight.
Sperm-specific protein kinase A catalytic subunit C® 2 orchestrates cAMP signaling for male fertility.
Proceedings of the National Academy of Sciences of the USA, 101(37):13483-13488, 2004. - 269
- P. Oberdoerffer,
C. Kanellopoulou, V. Heissmeyer,
C. Paeper, C. Borowski, I. Aifantis, A. Rao, and
K. Rajewsky.
Efficiency of RNA interference in the mouse hematopoietic system varies between cell types and developmental stages.
Molecular and Cellular Biology, 25(10):3896-3905, 2005. - 270
- A. O'Loghlen,
V. M. Gonzalez, D. Pineiro, M. I.
Perez-Morgado, M. Salinas, and M. E. Martin.
Identification and moleuclar characterization of Mnk1b, a splice variant of human MAP kinase-interacting kinase Mnk1.
Experimental Cell Research, 299:343-355, 2004. - 271
- A. J.
Paliga, D. R. Natale, and A. J. Watson.
p38 mitogen-activated protein kinase (MAPK) first regulates filamentous actin at the 8-16-cell stage during preimplantation development.
Biological Cell, 97:629-640, 2005. - 272
- R. D.
Palmiter, R. L. Brinster, R. E. Hammer, M. E. Trumbauer,
M. G. Rosenfeld, N. C. Birnberg, and R. M. Evans.
Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes.
Nature, 300:611-615, 1982. - 273
- Z.-Z. Pan,
Y. Devaux, and P. Ray.
Ribosomal S6 kinase as a mediator of keratinocyte growth factor-induced activation of Akt in epithelial cells.
Molecular Biology of the Cell, 15:3106-3113, 2004. - 274
- F. Park.
Lentiviral vectors: Are they the future of animal transgenesis.
Physiological Genomics, 2007.
DOI:10.1152. - 275
- J. L.
Parra, M. Buxade, and C. G. Proud.
Features of the catalytic domains and C termini of the MAP kinase signal-integrating kinases Mnk1 and Mnk2 determine their differing activities and regulatory properties.
The Journal of Biological Chemistry, 280(45):37623-33, 2005. - 276
- G. W.
Pearson and M. H. Cobb.
Cell condition-dependent regulation of ERK5 by cAMP.
The Journal of Biological Chemistry, 277(50):48094-48098, 2002. - 277
- G. W.
Pearson, S. Earnest, and M. H. Cobb.
Cyclic AMP selectively uncouples mitogen-activated protein kinase cascades from activating signals.
Molecular and Cellular Biology, 26(8):3039-3047, 2006. - 278
- P. Peraldi,
M. Frödin, J. Barnier, V. Calleja,
J.-C. Scimeca, C. Filloux, G. Calothy, and E. V.
Obberghen.
Regulation of the MAP kinase cascade in PC12 cells : B-Raf activates MEK-1 (MAP kinase or ERK kinase) and is inhibited by cAMP.
FEBS Letters, 357:290-296, 1995. - 279
- S. Peri,
J. Navarro, R. Amanchy, T. Kristiansen,
C. Jonnalagadda, V. Surendranath, V. Niranjan,
B. Muthusamy, T. Gandhi, M. Gronborg, N. Ibarrola,
N. Deshpande, K. Shanker, H. Shivashankar,
B. Rashmi, M. Ramya, Z. Zhao, K. Chandrika,
N. Padma, H. Harsha, A. Yatish, M. Kavitha,
M. Menezes, D. Choudhury, S. Suresh, N. Ghosh,
R. Saravana, S. Chandran, S. Krishna, M. Joy,
S. Anand, V. Madavan, A. Joseph, G. Wong,
W. Schiemann, S. Constantinescu, L. Huang,
R. Khosravi-Far, H. Steen, M. Tewari, S. Ghaffari,
G. Blobe, C. Dang, J. Garcia, J. Pevsner,
O. Jensen, P. Roepstorff, K. Deshpande,
A. Chinnaiyan, A. Hamosh, A. Chakravarti, and
A. Pandey.
Development of human protein reference database as an initial platform for approaching systems biology in humans.
Genome Research, 13(10):2363-2371, 2003. - 280
- L. Pianese,
L. Busino, I. D. Biase,
T. de Cristofaro, M. S. L. Casale,
P. Giuliano, A. Monticelli, M. Turano,
C. Criscuolo, A. Filla, S. Varrone, and S. Cocozza.
Up-regulation of c-Jun N-terminal kinase pathway in Friedreich's ataxia cells.
Human Molecular Genetics, 11(23):2989-2996, 2002. - 281
- S. Pichon,
M. Bryckaert, and E. Berrou.
Control of actin dynamics by p38 MAP kinase - Hsp27 distribution in the lamellipodium of smooth muscle cells.
Journal of Cell Science, 117(12):2569-2577, 2004. - 282
- R. Poirier,
S. Jacquot, C. Vaillend,
A. Soutthiphong, M. Libbey, S. Davis, S. Laroche,
A. Hanauer, H. Wzelzl, H.-P. Lipp, and D. Wolfer.
Deletion of the Coffin-Lowry syndrome gene Rsk2 in mice is associated with impaired spatial learning and reduced control of exploratory behavior.
Behavioral Genetics, 37:31-50, 2007. - 283
- G. W.
Porter, F. R. Khuri, and H. Fu.
Dynamic 14-3-3/client protein interactions integrate survival and apoptotic pathways.
Seminars in Cancer Biology, 16:193-202, 2006. - 284
- D. W.
Powell, M. J. Rane, B. A. Joughin, R. Kamukova, J.-H.
Hong, B. Tidor, W. L. Dean, W. M. Pierce, J. B.
Klein, M. B. Yaffe, and K. R. McLeish.
Proteomic identification of 14-3-3³ as a mitogen-activated protein kinase-activated protein kinase 2 substrate: role in dimer formation and ligand binding.
Molecular and Cellular Biology, 23(15):5376-5387, 2003. - 285
- W. H.
Press, S. A. Teukolsky, W. T. Vetterling, and B. P.
Flannery.
Numerical Recipes in C++, The art of Scientific Computing, chapter 15.7 Robust Estimation, pages 704-708.
Cambridge University Press, 2nd edition, 2003. - 286
- M. Qi,
M. Zhuo, B. S. Skalhegg, E. P. B. E. R. Kandel,
G. S. McKnight, and R. L. Idzerda.
Impaired hippocampal plasticity in mice lacking the C¯1 catalytic subunit of cAMP-dependent protein kinase.
Proceedings National Academy of Sciences of the USA, 93:1571-1576, 1996. - 287
- M. Raman.
Cre-loxP targeting.
http://www.geocities.com/mc_raman2004/Cre-lox.bmp, 2004. - 288
- M. Raman,
W. Chen, and M. Cobb.
Differential regulation and properties of mapks.
Oncogene, 26:3100-3112, 2007. - 289
- S. Ramboz,
R. Oosting, D. A. Amara, H. F. Kung,
P. Blier, M. Mendelsohn, J. J. Mann, D. Brunner,
and R. Hen.
Serotonin receptor 1A knockout: An animal model of anxiety-related disorder.
Proceedings of the National Academy of Science of the USA, 95:14476-14481, 1998. - 290
- L. Ramdas,
K. R. Coombes, K. Baggerly, L. Abruzzo,
W. E. Highsmith, T. Krogmann, S. R. Hamilton, and
W. Zhang.
Sources of nonlinearity in cDNA microarray expression measurements.
Genome Biol., 2(11), 2001. - 291
- D. Ramji
and P. Foka.
CCAAT/enhancer-binding proteins: structure, function and regulation.
Biochemical Journal, 365:561-575, 2002. - 292
- J. Randolph
and A. Waggoner.
Stability, specificity and fluorescence brightness of multiply-labeled fluorescent dna probes.
Nucleic Acid Res., 25(14):2923-2929, Jul 1997. - 293
- M. Rasar,
D. DeFranco, and S. R. Hammes.
Paxillin regulates steroid-triggered meiotic resumption in oocytes by enhancing an all-or-none positive feedback kinase loop.
The Journal of Biological Chemistry, 281:39455-39464, 2006. - 294
- K. Ravnskjaer,
H. Kester, Y. Liu, X. Zhang,
D. Lee, J. R. YatesIII, and M. Montminy.
Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression.
EMBO Journal, pages 1-10, 2007. - 295
- J. H.
Reiling, K. T. Doepfner, E. Hafen, and H. Stocker.
Diet-dependent effects of the Drosophila Mnk1/Mnk2 homolog Lk6 on growth via eIFeE.
Current Biology, 15:24-30, 2005. - 296
- H. C.
Reinhardt, A. S. Aslanian, J. A. Lees, and M. B. Yaffe.
p53-deficient cell rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage.
Cancer Cell, 11:175-189, 2007. - 297
- R. F.
Ren-Patterson, L. W. Cochran, A. Holmes, K.-P. Lesch,
B. Lu, and D. L. Murphy.
Gender-dependent modulation of brain monoamines and anxiety-like behaviors in mice with genetic serotonin transporter and BDNF deficiencies.
Cellular and Molecular Neurobiology, 26(4-6):755-780, 2006. - 298
- L. Reynolds
and R. Richards.
Can toxicogenomics provide information on the bioreactivity of diesel exhaust particles?
Toxicology, 165:145-152, 2001. - 299
- E. D.
Roberson, J. D. English, J. P. Adams, J. C. Selcher,
C. Kondratick, and J. D. Sweatt.
The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus.
The Journal of Neuroscience, 19(11):4337-4348, 1999. - 300
- A. Robinson-White
and C. A. Stratakis.
Protein kinase A signaling "Cross-Talk" with other pathways in endocrine cells.
Annals of the New York Academy of Sciences, 968:256-270, 2002. - 301
- R. G.
Roeder.
Transcriptional regulation and the role of diverse coactivators in animal cells.
FEBS Letters, 579:909-915, 2005. - 302
- D. Rogers,
E. Fisher, S. Brown, J. Peters,
A. Hunter, and J. Martin.
SHIRPA protocol: Behavioral and functional analysis of mouse phenotype assessment.
Mammalian Genome, 8(10):711-713, 1997. - 303
- N. Ronkina,
A. Kotlyarov, O. Dittrich-Breiholz,
M. Kracht, E. Hitti, K. Milarski, R. Askew,
S. Marusic, L.-L. Lin, M. Gaestel, and Telliez.
The mitogen-activated protein kinase (MAPK)-activated protien kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK.
Molecular and Cellular Biology, 27(1):170-181, 2007. - 304
- R. Roset,
L. Ortet, and G. Gol-Gomez.
Role of Bcl-2 family members on apoptosis: what we have learned from knock-out mice.
Frontiers in Bioscience, 12:4722-4730, 2007. - 305
- S. Rousseau,
N. Morrice, M. Peggie, D. G. Campbell,
M. Gaestel, and P. Cohen.
Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs.
The EMBO Journal, 21(23):6505-6514, 2002. - 306
- S. Rousseau,
M. Peggie, D. G. Campbell, A. R.
Nebreda, and P. Cohen.
Nogo-B is a new physiological substrate for MAPKAPK2.
Biochemistry Journal, 391:433-440, 2006. - 307
- P. P.
Roux and J. Blenis.
ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions.
Microbiology and Molecular Biology Reviews, 68(2):320-344, 2004. - 308
- T. Rulicke
and U. Hubscher.
Germ line transformation of mammals by pronuclear microinjection.
Experimental Physiology, 85:589-601, 2000. - 309
- P. J.
Russell.
iGenetics: A molecular approach.
Pearson/Benjamin Cummings, second edition, 2006. - 310
- R. Ryseck
and R. Bravo.
c-JUN, JUN B, and JUN D differ in their binding affinities to AP-1 and CRE consensus sequences: effect of FOS proteins.
Oncogene, 6:533-542, 1991. - 311
- S. D.
Sackett, J. T. Fulmer, J. R. Friedman, and K. H.
Kaestner.
Foxl1-Cre BAC transgenic mice: a new tool for gene ablation in the gastrointestinal mesenchyme.
Genesis, 45:518-522, 2007. - 312
- M. P.
Saelzler, C. C. Spackman, Y. Liu, L. C. Martinez,
J. P. Harris, and M. K. Abe.
ERK8 down-regulates transactivation of the glucocorticoid receptor through Hic-5.
The Journal of Biological Chemistry, 281(24):16821-16832, 2006. - 313
- F. Sananbenesi,
A. Fischer, C. Schrick, J. Spiess,
and J. Radulovic.
Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory.
The Journal of Neuroscience, 23(36):11436-11443, 2003. - 314
- G. Sanguinetti,
M. Milo, M. Rattray, and N. D.
Lawrence.
Accounting for probe-level noise in principal component analysis of microarray data.
Bioinformatics, 21(19):3748-3754, 2005. - 315
- P. J.
Santangelo, B. Nix, A. Tsourkas, and G. Bao.
Dual FRET molecular beacons for mRNA detection in living cells.
Nucleic Acids Research, 32(6):e57, 2004. - 316
- S. Sauma
and E. Friendman.
Increased expression of protein kinase C¯ activates ERK3.
The Journal of Biological Chemistry, 271(19):11422-11426, 1996. - 317
- T. Sawa
and L. Ohno-Machado.
A neural network-based similarity index for clustering DNA microarray data.
Computers in Biology and Medicine, 33(1):1-15, 2003. - 318
- M. Saxena,
S. W. K. Tasken, and T. Mustelin.
Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase.
Nature Cell Biology, 1:305-311, 1999. - 319
- E. Schneider,
M. Weiss, W. du, G. Leder,
K. Buttenschön, U. Liener, and U. Brückner.
MAPkinase gene expression, as determined by microarray analysis, distinguishes uncomplicated from complicated reconstitution after major surgical trauma.
Annals of the New York Academy of Sciences, 1090:429-444, 2006. - 320
- S. Schumacher,
K. Laas, S. Kant, Y. shi,
A. Kotlyarov, and M. Gaestel.
Scaffolding by ERK3 regulates MK5 in development.
The EMBO Journal, 23(24):4770-9, 2004. - 321
- M. Schäferling
and S. Nagl.
Optical technologies for the read out and quality control of DNA and protein microarrays.
Analytical and Bioanalytical Chemistry, 385(3):500-517, 2006. - 322
- C. Schäffer,
S. Ross, M. Bragado,
G. Groblewski, S. Ernst, and J. Williams.
A role for the p38 mitogen-activated protein kinase/Hsp 27 pathway in cholecystokinin-induced changes in the actin cytoskeleton in rat pancreatic acini.
Journal of Biological Chemistry, 273:24173-24180, 1998. - 323
- S. Seino
and T. Shibasaki.
PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis.
Physiological Reviews, 85:1303-1342, 2005. - 324
- O. M.
Seternes, B. Johansen, B. Hegge, M. Johannessen,
S. M. Keyse, and U. Moens.
Both binding and activation of p38 mitogen-activated protein kinase (MAPK) play essential roles in regulation of the nucleocytoplasmic distribution of MAPK-activated protein kinase 5 by cellular stress.
Molecular and Cellular Biology, 22(20):6931-6945, 2002. - 325
- O. M.
Seternes, T. Mikalsen, B. Johansen, E. Michaelsen,
C. G. Armstrong, N. A. Morrice, B. Turgeon,
S. Meloche, U. Moens, and S. M. Keyse.
Activation of MK5/PRAK by the atypcial MAP kinase ERK3 defines a novel signal transduction pathway.
The EMBO Journal, 23:4780-4791, 2004. - 326
- J. W.
Shay and W. E. Wright.
Senescence and immortalization: role of telomeres and telomerase.
Carcinogenesis, 25(5):867-874, 2005. - 327
- M. Shelly,
L. Cancedda, S. Heilshorn, G. Sumbre,
and M. ming Poo.
LKB1/STRAD promotes axon initiation during neuronal polarization.
Cell, 129:565-577, 2007. - 328
- Y. Shi,
A. Kotlyarov, K. Laas, A. D. Gruber, E. Butt,
K. Marcus, H. E. Meyer, A. Friedrich, H.-D. Volk, and
M. Gaestel.
Elimination of protein kinases MK5/PRAK activity by targeted homologous recombination.
Molecular and cellular biology, 23(21):7732-7741, 2003. - 329
- A. Shimomura,
Y. Okamoto, Y. Hirata, M. Kobayashi,
K. Kawakami, K. Kiuchi, T. Wakabayashi, and
M. Hagiwara.
Dominant negative ATF1 blocks cyclic AMP-induced neurite outgrowth in PC12D cells.
Journal of Neurochemistry, 70:1029-1034, 1998. - 330
- K. Shiroto,
H. Otani, F. Yamamoto, C.-K. Huang,
N. Maulik, and D. K. Das.
MK2-/- gene knockout mouse hearts carry anti-apoptotic signal and are resistant to ischemia reperfusion injury.
Journal of Molecular and Cellular Cardiology, 38:93-97, 2005. - 331
- S. Shoji,
K. Titani, J. Demaille, and E. Fischer.
Sequence of two phosphorylated sites in the catalytic subunit of bovine cardiac muscle adenosine 3':5'-monophosphate-dependent protein kinase.
The Journal of Biological Chemistry, 254:6211-6214, 1979. - 332
- M. O.
Showers and R. A. Maurer.
A cloned cDNA encodes an alternate form of the catalytic subunit of cAMP-dependent protein kinase.
The Journal of Biological Chemistry, 261(35):16288-16291, 1986. - 333
- E. Silverman,
M. Frödin, S. Gammeltoft, and
J. L. Maller.
Activation of p90RSK is sufficient for differentiation of PC12 cells.
Molecular and Cellular biology, 24(24):10573-10583, 2004. - 334
- C. B.
Sindreu, Z. S. Scheiner, and D. R. Storm.
Ca -stimulated adenylyl cyclases
regulate ERK-dependent activation of MSK1 during fear conditioning.
-stimulated adenylyl cyclases
regulate ERK-dependent activation of MSK1 during fear conditioning.
Neuron, 53:79-89, 2007. - 335
- G. Sithanandam,
F. Latif, F. Duh, R. Bernal,
U. Smola, H. Li, I. Kuzmin, V. Wixler,
L. Geil, and S. Shrestha.
3pK, a new mitogen-activated protein kinase-activated protein kinase located in the small cell lung cancer tumor suppressor gene region.
Molecular and Cellular Biology, 16:868-876, 1996. - 336
- B. S.
Skalhegg, Y. Huang, T. Su, R. L. Idzerda, G. S.
McKnight, and K. A. Burton.
Mutation of the C® subunit of PKA leads to growth retardation and sperm dysfunction.
Molecular Endocrinology, 16(3):630-639, 2002. - 337
- B. S.
Skålhegg and K. Tasken.
Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA.
Frontiers in Bioscience, 5:678-693, 2000. - 338
- K. Slentz-Kesler,
J. T. Moore, M. Lombard,
J. Zhang, R. Hollingsworth, and M. P. Weiner.
Identification of the human Mnk2 gene (MKNK2) trhough protein interaction with the estrogen receptor .
Genomics, 69:63-71, 2000. - 339
- H. A.
Slotten, M. Kalinichev, J. J. Hagan, C. A. Marsden, and
K. C. F. Fone.
Long-lasting changes in behavioural and neuroendocrine indices in the rat following neonatal maternal separation: Gender-dependent effects.
Brain Research, 1097:123-132, 2006. - 340
- K. J.
Smith, P. S. Carter, A. Bridges, P. Horrocks,
C. Lewis, G. Pettman, A. Clarke, M. Brown,
J. Hughes, M. Wilkinson, B. Bax, and A. Reith.
The structure of MSK1 reveals a novel autoinhibitory conformation for a dual kinase protein.
Structure, 12:1067-1077, 2004. - 341
- G. K.
Smyth and T. P. Speed.
Normalization of cDNA microarray data.
Methods, 31:265-273, 2003. - 342
- A. Soloaga,
S. Thomson, G. R. Wiggin,
N. Rampersaud, M. H. Dyson, C. A. Hazzalin, L. C.
Mahadevan, and J. S. C. Arthur.
MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14.
The EMBO Journal, 22(11):2788-2797, 2003. - 343
- F. Song
and R. Goodman.
OsBIMK1, a rice MAP kinase gene involved in disease resistance responses.
Planta, 215:997-1005, 2002. - 344
- R. K.
Srivastava, A. R. Srivastava, P. Seth, S. Agrawal, and
Y. S. Cho-Chung.
Growth arrest and induction of apoptosis in breast cancer cells by antisense depleation of protein kinase A-RI® subunit: p53-independent mechanism of action.
Molecular and Cellular Biochemistry, 195:25-36, 1999. - 345
- R. A.
Steiner, J. G. Hohmann, A. Holmes, C. C. Wrenn,
G. Cadd, A. Jureus, D. K. Clifton, M. Luo,
M. Gutshall, S. Y. Ma, E. J. Mufson, and J. N.
Crawley.
Galanin transgenic mice display cognitive and neurochemical deficits characteristic of Alzheimer's disease.
Proceedings of the National Academy of Sciences of the USA, 98(7):4184-4189, 2001. - 346
- D. Stokoe,
D. Campbell, S. Nakielny, H. Hidaka,
S. Leevers, C. Marshall, and P. Cohen.
MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase.
The EMBO Journal, 11:3985-3994, 1992. - 347
- P. J.
Stork and J. M. Schmitt.
Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation.
Trends in Cell Biology, 12(6):258-266, 2002. - 348
- K. Struhl.
Fundamentally different logic of gene regulation in eukaryotes and prokaryotes.
Cell, 98:1-4, 1999. - 349
- T. Sudo,
K. Kawai, H. Matsuzaki, and H. Osada.
p38 mitogen-activated protein kinase plays a key role in regulating MAPKAPK2 expression.
Biochemical and Biophysical Research Communications, 337:415-421, 2005. - 350
- M. Sun,
Y. Wei, L. Yao, J. Xie, X. Chen, H. Wang,
J. Jiang, and J. Gu.
Identification of extracellular signal-regulated kinase 3 as a new interaction partner of cyclin d3.
Biochemical and Biophysical Research Communications, 340:209-214, 2006. - 351
- P. Sun,
N. Yoshizuka, L. New, B. A. Moser, Y. Li,
R. Liao, C. Xie, J. Chen, Q. Deng, M. Yamout,
M.-Q. Dong, C. G. Frangou, J. R. YatesIII, P. E. Wright,
and J. Han.
PRAK is essential for ras-induced senescence and tumor suppression.
Cell, 128:295-308, 2007. - 352
- J. D.
Sweat.
The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory.
Journal of Neurochemistry, 76:1-10, 2001. - 353
- G. Szapiro,
M. R. Vienna, J. L. McGaugh, J. H.
Medina, and I. Izquierdo.
The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear.
Hippocampus, 13:53-58, 2003. - 354
- H. Szutorisz,
N. Dillon, and L. Tora.
The role of enhancers as centres for general transcription factor recruitment.
TRENDS in Biochemical Science, 30(11):593-599, 2005. - 355
- H. Tak,
E. Jang, S. B. Kim, J. Park, J. Suk, Y. S.
Yoon, J. K. Ahn, J.-H. Lee, and C. O. Joe.
14-3-3epsilon inhibits MK5-mediated cell migration by disrupting F-actin polymerization.
Cellular Signaling, 2007. - 356
- G. Takaesu,
J.-S. Kang, G.-U. Bae, M.-J. Yi, C. M. Lee,
E. P. Reddy, and R. S. Krauss.
Activation of p38®/¯ MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo.
Journal of Cell Biology, 175(3):383-388, 2006. - 357
- K. Tamura,
T. Sudo, U. Senftleben, A. M. Dadak,
R. Johnson, and M. Karin.
Requirement for p38® in erythropoietin expression: a role for stress kinases in erythropoiesis.
Cell, 102:221-231, 2000. - 358
- T.-T. Tan and
L. M. Coussens.
Humoral immunity, inflammation and cancer.
Current Opinion in Immunology, 19:209-216, 2007. - 359
- T. Tanoue,
R. Maeda, M. Adachi, and E. Nishida.
Identification of a docking groove on ERK and p38 MAPK that regulates the specificity of docking interactions.
The EMBO Journal, 20(3):466-479, 2001. - 360
- D. Tantin,
C. Schild-Poulter, V. Wang,
R. Haché, and P. Sharp.
The octamer binding transcription factor Oct-1 is a stress sensor.
Cancer Research, 65:10750-10758, 2005. - 361
- K. Taskén
and E. M. Aandahl.
Localized effects of cAMP mediated by distinct routes of protein kinase A.
Physiological Reviews, 84:137-167, 2004. - 362
- L. Taupenot,
K. L. Harper, and D. T. O'Connor.
Mechanisms of disease : The chromogranin-secretogranin family.
The New England Journal of Medicine, 348:1134-49, 2003. - 363
- S. S.
Taylor, C. Kim, D. Vigil, N. M. Haste, J. Yang,
J. Wu, and G. S. Anand.
Dynamics of signaling by PKA.
Biochimica et Biophysica Acta, 1754:25-37, 2005. - 364
- S. S.
Taylor, E. Radzio-Andzelm, Madhusudan, X. Cheng, L. T.
Eyck, and N. Narayana.
Catalytic subunit of cyclic AMP-dependent protein kinase: structure and dynamics of the active site cleft.
Pharmacol. Ther., 82(2-3):133-141, 1999. - 365
- K. Tenbrock,
Y.-T. Juang, V. Kyttaris, and G. Tsokos.
Altered signal transduction in SLE T cells.
Rheumatology, 46:1525-1530, 2007. - 366
- G. Thiel,
V. A. Sarraj, C. Vinson, L. Stefano, and
K. Bach.
Role of basic region leucine zipper transcription factors cyclic AMP response element binding protein (CREB), CREB2, activating transcription factor 2 and CAAT/enhancer binding protein alpha in cyclic AMP response element-mediated transcription.
Journal of Neurochemistry, 92:321-336, 2005. - 367
- G. M.
Thomas, G. R. Rumbaugh, D. B. Harrar, and R. L. Huganir.
Ribosomal S6 kinase 2 interacts with and phosphorylates PDZ domain-containing proteins and regulates AMPA receptor transmission.
Proceedings of the National Academy of Science of the USA, 102(42):15006-15011, 2005. - 368
- M. Thomas
and C. Chiang.
The general transcription machinery and general cofactors.
Critical Reviews in Biochemistry and Molecular Biology, 41(3):105-178, 2006. - 369
- T. Thuraisingam,
Y. Z. Xu, J. Moisan, C. Lachance,
J. Garnon, S. D. Marco, M. Gaestel, and
D. Radzioch.
Distinct role of MAPKAPK-2 in the regulation of TNF gene expression by Toll-like receptor 7 and 9 ligands.
Molecular Immunology, 44:3482-3491, 2007. - 370
- G. Thyssen,
T.-H. Li, L. Lehmann, M. Zhuo,
M. Sharma, and Z. Sun.
LZTS2 is a novel -catenin interacting
protein and regulates the nuclear export of -catenin.
Molecular and Cellular Biology, 2006. - 371
- M. Tiainen,
A. Ylikorkala, and T. P. Mäkelä.
Growth suppression by Lkb1 is mediated by a G1 cell cycle arrest.
Proceedings of the National Academy of Sciences of USA, 96:9248-9251, 1999. - 372
- A. Tietz,
A. Malo, J. Diebold, A. Kotlyarov,
A. Herbst, F. Kolligs, B. Brandt-Nedelev,
W. Halangk, M. Gaestel, B. Göke, and
C. Schäfer.
Gene deletion of MK2 inhibits TNF® and IL-6 and protects against cerulein-induced pancreatitis.
American Journal of Physiology Gastrointestinal and Liver Physiology, 290(6):G1298-306, 2006. - 373
- C. Tinti,
C. Yang, H. Seo, B. Conti, C. Kim,
T. H. Joh, and K.-S. Kim.
Structure/function relationship of the cAMP response element in tyrosine hydroxylase gene transcription.
The Journal of Biological Chemistry, 272(31):19158-19164, 1997. - 374
- K. Toska,
R. Kleppe, C. G. Armstrong, N. A.
Morrice, P. Cohen, and J. Haavik.
Regulation of tyrosine hydroxylase by stress-activated protein kinases.
Journal of Neurochemistry, 83:775-783, 2002. - 375
- K. Toska,
R. Kleppe, P. Cohen, and J. Haavik.
Phosphorylation of tyrosine hydroxylase in isolated mice adrenal glands.
Annals of the New York Academy of Science, 971:66-68, 2002. - 376
- Z. Travnickova-Bendova,
N. Cermakian, S. M. Reppert, and
P. Sassone-Corsi.
Bimodal regulation of mPeriod promotors by CREB-dependent signaling and CLOCK/BMAL1 activity.
Proceedings of the National Academy of Sciences, 99(11):7728-7733, 2002. - 377
- P. Trifilieff,
C. Herry, P. Vanhoutte, J. Caboche,
A. Desmedt, G. Riedel, N. Mons, and J. Micheau.
Foreground contextual fear memory consolidation requires two independent phases of hippocampal ERK/CREB activation.
Learning and Memory, 13:349-358, 2006. - 378
- F. Tronche,
C. Kellendonk, O. Kretz, P. Gass,
K. Anlag, P. C. Orban, R. Bock, R. Klein, and
G. Schütz.
Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety.
Nature Genetics, 23:99-103, 1999. - 379
- B. Turgeon,
M. K. Saba-El-Leil, and S. Meloche.
Cloning and characterization of mouse extracellular-signal-regulated protein kinase 3 as a unique gene product of 100kDa.
Biochemistry Journal, 346:169-175, 2000. - 380
- A. Turjanski,
J. Vaque, and J. Gutkind.
MAP kinases and the control of nuclear events.
Oncogene, 26:3240-3253, 2007. - 381
- T. Ueda,
R. Watanabe-Fukunaga, H. Fukuyama, S. Nagata, and
R. Fukunaga.
Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development.
Molecular and Cellular Biology, 24(15):6539-6549, 2004. - 382
- K. W.
Underwood, K. D. Parris, E. Federico, L. Mosyak,
R. M. Czerzinski, T. Shane, M. Taylor, K. Svenson,
Y. Liu, C.-L. Hsiao, S. Wolfrom, M. Maguire,
K. Malakian, J.-B. Telliez, L.-L. Lin, R. W. Kriz,
J. Seehre, W. S. Somers, and M. L. Stahl.
Catalytically active MAPKAP kinase 2 structures in complex with staurosporine and ADP reveal differences with the autoinhibited enzyme.
Structure, 11:627-636, 2003. - 383
- M. VanDijk,
L. Peltenburg, and C. Murre.
Hox gene products modulate the DNA binding activity of Pbx1 and Pbx2.
Mechanisms of Development, 52:99-108, 1995. - 384
- Y. Vera,
T. Dai, A. P. S. Hikim, Y. Lue, E. C. Salido,
R. S. Swerdloff, and P. H. Yen.
Deleted in Azoospermia Associated Protein 1 shuttles between nucleus and cytoplasm during normal germ cell maturation.
Journal of Andrology, 23(5):622-628, 2002. - 385
- A. Vertii,
C. Hakim, A. Kotlyarov, and M. Gaestel.
Analysis of properties of small heat shock protein Hsp25 in MAPK-activated protein kinase 2 (MK)-deficient cells.
The Journal of Biological Chemistry, 37(37):2699-26975, 2006. - 386
- L. J.
Vician, G. Xu, W. Liu, J. D. Feldman, H. B.
Machado, and H. R. Herschman.
MAPKAP kinase-2 is a primary response gene induced by depolarization in PC12 cells in brain.
Journal of Neuroscience Research, 78:315-328, 2004. - 387
- D. Vignjevic,
S. Fre, D. Louvard, and S. Robine.
Conditional mouse models of cancer.
Handbook of Experimental Pharmacology, 178:263-87, 2007. - 388
- J. Villard.
Transcription regulation and human diseases.
Swiss Med WKLY, 134:571-578, 2004. - 389
- N. Vo,
M. Klein, O.Varlamova, D. Keller, T. Yamamoto,
R. Goodman, and S. Impey.
A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis.
Proceedings of the National Academy of Sciences of the USA, 102:16426-16431, 2005. - 390
- J. W.
Voncken, H. Niessen, B. Neufeld, U. Rennefahrt,
V. Dahlmans, N. Kubben, B. Holzer, S. Ludwig, and
U. R. Rapp.
MAPKAP kinase 3pK phosphorylates and regulates chromatin-association of the polycomb-group protein Bmi1.
Journal of Biological Chemistry, 280(7):5178-87, 2004. - 391
- T. Wada
and J. M. Penninger.
Mitogen-activated protein kinases in apoptosis regulation.
Oncogene, 23:2838-2849, 2004. - 392
- H. M.
Wain, E. A. Bruford, R. C. Lovering, M. J. Lush,
M. W. Wright, and S. Povey.
Guidelines for Human Gene Nomenclature.
Genomics, 79(4):464-470, 2002. - 393
- H. A.
Wallace, F. Marques-Kranc, M. Richardson,
F. Luna-Crespo, J. A. Sharpe, J. Hughes, W. G.
Wood, D. R. Higgs, and A. J. Smith.
Manipulating the mouse genome to engineer precise functional syntenic replacements with human sequence.
Cell, 128:197-209, 2007. - 394
- R. Waltereit
and M. Weller.
Signaling from cAMP/PKA to MAPK and synaptic plasticity.
Molecular Neurobiology, 27(1):99-106, 2003. - 395
- X. Wang,
L. Xu, H. Wang, P. R. Young, and M. G. adn Giora
Z. Feuerstein.
Mitogen-activated protein kinase-activated protein (MAPKAPK) kinase 2 deficiency protects brain from ischemic injury in mice.
The Journal of Biological Chemistry, 277(46):43968-43972, 2002. - 396
- A. White,
C. A. Pargellis, J. M. Studts, B. G.
Werneburg, and B. T. FarmerII.
Molecular basis of MAPK-activated protein kinase 2:p38 assembly.
Proceedings of the National Academy of Sciences, 104(15):6353-8, 2007. - 397
- A. J.
Whitmarsh.
Regulation of gene transcription by mitogen-activated protein kinase signaling pathways.
Biochimica et Biophysica Acta, 1773(8):1285-1298, 2007. - 398
- G. R.
Wiggin, A. Soloaga, J. M. Foster, V. Murray-Tait,
P. Cohen, and J. S. C. Arthur.
MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts.
Molecular and Cellular Biology, 22(8):2871-2881, 2002. - 399
- E. Wingender,
X. Chen, R. Hehl, H. Karas,
I. Liebich, T. Matys, T. Meinhardt, M. Pruss,
I. Reuter, and F. Schacherer.
Transfac: an integrated system for gene expression regulation.
Nucleic Acids Research, 28:316-319, 2000. - 400
- A. Woods,
S. R. Johnstone, K. Dickerson, F. C.
Leiper, L. G. Fryer, D. Neumann, U. Schlattner,
T. W. M. Carlson, and D. Carlong.
LKB1 is the upstream kinase in the AMP-activated protein kinase cascade.
Current Biology, 13:2004-2008, 2003. - 401
- J. Worch,
L. Tickenbrock, J. Schwäble,
B. Steffen, T. Cauvet, B. Mlody, H. Buerger,
H. P. Koeffler, W. E. Berdel, H. Serve, and
C. Müller-Tidow.
The serine-threonine kinase MNK1 is post-translationally stabilized by PML-RAR® .
Oncogene, 23:9162-9172, 2004. - 402
- J. Workman.
Nucleosome displacement in transcription.
Genes and Development, 20(15):2009-2017, 2006. - 403
- J. Wu
and R. Janknecht.
Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A.
The Journal of Biological Chemistry, 277(45):42669-42679, 2002. - 404
- J.-P. Wu,
J. Wang, A. Abeywardane, D. Andersen, M. Emmanuel,
E. Gautschi, D. R. Goldberg, M. A. Kashem,
S. Lukas, W. Mao, L. Martin, T. Morwick,
N. Moss, C. Pargellis, U. R. Patel, L. Patnaude,
G. W. Peet, D. Skow, R. J. Snow, Y. Ward,
B. Werneburg, and A. White.
The discovery of carboline analogs as potent MAPKAP-K2 inhibitors.
Bioorganic and Medicinal Chemistry Letters, 2007. - 405
- R. Wu,
H. Kausar, P. Johnson, D. Montoya-Durango,
M. Merchant, and M. Rane.
Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex.
The Journal of Biological Chemistry, 282:21598-21608, 2007. - 406
- X.-G. Xia,
H. Zhou, and Z. Xu.
Transgenic RNAi: accelerating and expanding reverse genetics in mammals.
Transgenic Research, 15:271-275, 2006. - 407
- Y. Xiao
and P. E. Barker.
Semiconductor nanocrystal probes for human metaphase chromosomes.
Nucleic Acids Research, 32(3):e28, 2004. - 408
- L. Yan,
J. Carr, P. A. PR, V. Murry-Tait, C. Thompson, and
J. Arthur.
Knockout of ERK5 causes multiple defects in placental and embryonic development.
BMC Developmental Biology, 3:11, 2003. - 409
- Q. Yan,
E. Reed, X. Zhong, K. Thornton, Y. Guo, and
J. Yu.
MZF1 possesses a repressively regulatory function in ERCC1 expression.
Biochemical Pharmacology, 71:761-771, 2006. - 410
- J. Yao,
J. D. Erickson, and L. B. Hersh.
Protein kinase A affects trafficking of the vesicular monoamine transporters in pc12 cells.
Traffic, 5:1006-1016, 2004. - 411
- F. Yates.
Contingency table involving small numbers and the Â2 test.
Journal of the Royal Statistical Society, (Supplement) 1:217-235, 1934. - 412
- H. G.
Yntema, B. van den Helm, J. Kissing, G. van
Duijnhoven, F. Poppelaars, J. Chelly, C. Moraine, J.-P.
Fryns, B. C. J. Hamel, H. Heilbronner, H.-J. Pander,
H. G. Brunner, H.-H. Ropers, F. P. Cremers, and H. van
Bokhoven.
A novel ribosomal-S6-kinase (RSK';RPS6KA6) is commonly deleted in patients with complex X-linked mental retardation.
Genomics, 62:332-343, 1999. - 413
- K. Yoshioka.
Scaffold proteins in mammalian MAP kinase cascades.
Journal of Biochemistry, 135(6):1-5, 2004. - 414
- P. Young,
P. McDonnell, D. Dunnington, A. Hand,
J. Laydon, and J. Lee.
Pyridinyl imidazoles inhibit IL-1 and TNF production at the protein level.
Agents Actions, 39:C67-C69, 1993. - 415
- V. Zachariou,
D. H. Brunzell, J. Hawes, D. R.
Stedman, T. Bartfai, R. A. S. an David Wynick,
Ülo Langel, and M. R. Picciotto.
The neuropeptide galanin modulates behavioral and neurochemical signs of opiate withdrawal.
Proceedings of the National Academy of Science of the USA, 100(15):9028-9033, 2003. - 416
- S. Zadelaar,
R. Kleemann, L. Verschuren, J. deVries
VanderWeij, J. vanderHoom, H. M. Princen, and
T. Kooistra.
Mouse models for atherosclerosis and pharmaceutical modifiers.
Arteriosclerosis, Thrombosis and Vascular Biology, 27:1706-1721, 2007. - 417
- V. Zakowski,
G. Keramas, K. Kilian, U. R. Rapp, and
S. Ludwig.
Mitogen-activated 3p kinase is active in the nucleus.
Experimental Cell Research, 299:101-109, 2004. - 418
- R. Zhang,
S. Murakami, F. Coustry, Y. Wang, and
B. de Crombrugghe.
Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation.
Proceedings of the National Academy of Sciences of the USA, 103(2):365-370, 2006. - 419
- X. Zhang,
D. T. Odom, S.-H. Koo, M. D. Conkright,
C. Canettieri, J. Best, H. Chen, R. Jenner,
E. Herbolsheimer, E. Jacobsen, S. Kadam, J. R.
Ecker, B. Emerson, J. B. Hogenesch, T. Unterman,
R. A. Young, and M. Montminy.
Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation and target gene activation in human tissues.
Proceedings of the National Academy of Science, 102:4459-4464, 2005. - 420
- Y. Zhao,
C. Bjorbak, and D. E. Moller.
Regulation and interaction of pp90RSK isoforms with mitogen-activated protein kinases.
The Journal of Biological Chemistry, 271(47):29772-29779, 1996. - 421
- C. Zheng,
Z. Lin, Z. J. Zhao, Y. Yang, H. Niu,
and X. Shen.
MAPK-activated protein kinase-2(MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt and Hsp27.
The Journal of Biological Chemistry, 281(48):37215-37226, 2006. - 422
- G. Zhou,
Z. Q. Bao, and J. E. Dixon.
Components of a new human protein kinase signal transduction pathway.
The Journal of Biological Chemistry, 270(21):12665-12669, 1995. - 423
- C.-B. Zhu,
A. M. Carneiro, W. R. Dostmann, W. A. Hewlett, and
R. D. Blakely.
p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2a-dependent process.
The Journal of Biological Chemistry, 280(16):15649-15658, 2005. - 424
- C.-B. Zhu,
W. A. Hewlett, I. Feoktistov, I. Biaggioni, and
R. D. Blakely.
Adenosine receptor, protein kinase G, and p38 mitogen-activated protein kinase-dependent up-regulation of serotonin transporters involves both transporter trafficking and activation.
Molecular Pharmacology, 65(6):1462-1474, 2004. - 425
- B. Zimmermann,
J. A. Chiorini, Y. Ma, R. M. Kotin,
and F. W. Herberg.
PrKX is a novel catalytic subunit of the cAMP-dependent protein kinase regulated by the regulatory subunit type I.
The Journal of Biological Chemistry, 274(9):5370-5378, 1999. - 426
- E. Åberg,
M. Perander, B. Johansen,
C. Julien, S. Meloche, S. M. Keyse, and O.-M. Seternes.
Regulation of the MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAP kinase ERK4/MAPK4.
Journal of Biological Chemistry, 281(46):35499-510, 2006.
Index
- 14-3-3
- 2.3.1.1 | 2.3.4.3.3 | 2.3.4.3.5 | 2.3.4.3.5 | 2.4.2.5
- actin
- 2.3.4.3.4 | 2.4.2.5 | A.
- activation loop
- 2. | 2.2 | 2.2.3 | 2.2.4 | 2.2.4 | 2.3.2 | 2.3.2.1 | 2.4.1
- adaptors
- 1.2 | 2. | 2.
- AGC
- 1.1.1 | 2.3.1.1 | 3.1
- AKAP
- 3.3 | 3.3 | 3.3 | 3.3 | 3.5.1 | 3.5.2
- anisomycin
- 2.3.4.3.5 | 2.4.2.1 | A.
- anxiety-related
- Abstract | Summary | Summary | Conclusion | Current | Abstract | Background | Differences | Differences | Light-dark | Discussion | Difference | Anxiety-related | Anxiety-related
- arthritis
- 1.1.2 | 2.3.4.3.7
- atypical MAPKs
- 2.2.5
- AU-rich elements (ARE)
- 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.6 | 1. | 1.
- auto-inhibitory domain
- 2.1.1 | 2.1.1 | 2.1.1 | 2.3.4.2.1 | 2.3.4.2.2 | 2.4.1 | 3.2
- autophosphorylation
- 1.2 | ERK7 | 2.3.1.1 | 2.3.2
- B-Raf
- 1.1.2 | 1.1.2 | 2.2.1 | 2.2.1 | 3.5 | Background
- BJ cells
- 2.4.2.4
- blastocyst
- 5.2.1.2 | 5.2.1.2 | 5.2.1.2 | 5. | 5. | MK5
- BRET
- 5.2.2
- C2C12 cells
- 3.5
- CAAT-box
- 4.2 | 4.2
- carboline
- 1.1.2
- catalepsy
- 3.4
- catalytic site
- 2.1.1 | 2.1.1 | 2.1.1 | 2.3.4.2.2 | 2.4.1
- CD domain
- 2.1.2
- cdc42
- 1.2.2
- cell cycle
- 1.2.2 | ERK3 | 2.3.1.2 | 2.3.4.3.5 | 2.4.2.5 | 3.2 | 1. | 3.
- chromatin
- 2.3.4.3.6 | 4. | 4. | 4. | 4.1 | 4.1 | 4.1 | 4.2
- CNS
- 2.3.1 | 2.3.1
- conformational change
- 1. | 1. | 2.2.4 | 2.3.2.1 | 2.3.2.1 | 2.3.3 | 3.2 | 3.3
- consensus sequence
- 2.3.4.2.1 | 2.4.1 | 2.4.1 | 4.2 | 4.2 | 4.2 | 4.2 | 1. | The | Figures | Figures
- COS cells
- 2.3.4.1 | Construction | Cell | Transient | The | Discussion | Figures | Figures
- CPK
- 2.1.1 | 2.1.1
- Cre/loxP
- 5.2.1.4.1 | 5.2.1.4.1 | 5.2.1.4.1 | 5.2.1.4.1 | 5.2.1.4.1
- CREB
- Abstract | 2.3 | 2.3.1.1 | 2.3.2.2 | 2.3.4.1 | 2.3.4.3.6 | 3.3 | 3.4 | 3.5.1 | 3.5.2 | 3.5.2 | Summary | 4.2 | 4.2 | 4.2 | Abstract | Materials | Materials | Plasmids | Plasmids | Electrophoresis | Small | CREB | CREB | MK5 | MK5 | The | The | The | The | The | The | The | MK5 | The | Discussion | Discussion | Discussion | Discussion | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures
- Crm1
- ERK3 | 2.3.4.3.2 | 2.4.2.2 | 2.4.2.3
- crosstalk
- 3.5 | 3.5 | 3.5.2
- cycloheximide
- 2.2.3 | A.
- cytokine
- 2. | 2.2.1 | 2.2.3 | 2.2.3 | 2.2.4 | 2.3.4.3.3 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.7 | 2.3.4.3.7 | 2.3.4.3.7 | 2.3.4.3.7 | 2.4.3 | 2.4.3 | 3.4 | Introduction
- cytoskeleton
- Abstract | 1.2.2 | 3. | 4. | 4.1 | 4.2 | 4.3 | A.
- development
- 2.2.4 | ERK3 | ERK3 | 2.3.1 | 2.3.1 | 2.3.1.2 | 2.3.3.2 | 2.3.4.3.7 | 2.4.2.3 | 2.4.3 | 5.2.1.6.1 | 3. | 3. | 4.1 | Background | General | Difference | MK5
- developmental
- Discussion
- differentiation
- Abstract | 2.2.1 | 2.2.2 | 2.2.4 | ERK3 | ERK3 | 2.3.1.2 | 2.3.1.2 | 2.3.3.2 | 2.3.4.3.6 | 2.3.4.3.6 | 3.2 | 5.2.1.6 | 6 | Summary | 4. | 4.3 | 4.3 | Current | Abstract | Background | Abstract | Discussion
- DKO
- 2.3.3.2 | 2.3.4.3.6 | 2.3.4.3.7 | 2.3.4.3.7 | 2.3.4.3.7 | 2.4.3 | 2.4.3 | Anxiety-related
- DMBA
- 2.4.3 | 3.4 | 3. | Background
- docking
- 2.1.2 | 2.1.2 | 2.1.2 | 2.2 | 2.3.2 | 2.3.3 | 2.3.3 | 2.3.4.2.1 | 2.3.4.2.2 | 2.4.2.1 | 2.4.2.1 | 3.2 | 3.2 | 3.2 | 3.3 | 4.3
- dopamine
- 2.4.2.7.3 | 2.4.2.7.3 | 3.5.1 | 5.2.1.6 | Context | Current | Current | Current | Activity | Activity
- Drosophila
- 1.1.1 | 2.3.3.1 | 2.3.3.2 | 2.4 | 4.2 | 4.2 | 4.3 | A.
- enhancers
- 4.2 | 4.2
- Epac
- 1.2.2 | 3.5.1 | 3.5.1
- EPM
- Abstract | List | Summary | 5. | 1. | Background | Behavioural | Behavioural | Image | Image | Image | Statistical | Differences | Differences | Differences | Differences | Differences | Differences | Differences | Differences | Mouse | Mouse | Mouse | Detailed | Detailed | Light-dark | Difference | Difference | Difference | Anxiety-related | Anxiety-related | Anxiety-related | Anxiety-related | Activity | Activity
- ER81
- 2.3.2.2 | 2.3.2.2 | 2.3.2.2 | 2.3.4.3.6 | 3.5.2 | 3.5.2 | 3.5.2 | 3.5.2
- ERK
- no title
- ERK3
- 1.1.1 | ERK3 | ERK3 | ERK3 | ERK3 | ERK3 | ERK3 | ERK3 | ERK4 | ERK7 | 2.4.2.2 | 2.4.2.2 | 2.4.2.2 | 2.4.2.2 | 2.4.2.3 | 2.4.2.3 | 2.4.3 | 4.1 | 1. | 3. | 3. | 4.3 | 1. | Introduction | Discussion | Discussion
- ERK4
- 1.1.1 | ERK3 | ERK3 | ERK4 | ERK4 | ERK4 | 2.4.2.3 | 2.4.2.3 | 1. | 1.
- ERK6
- 2.2.4
- ES cells
- 5.2.1.2 | 5.2.1.2 | 5.2.1.4.3 | 5.2.1.6.1 | 5.
- FISH
- 5.2.2 | 5.
- FLP/FRT
- 5.2.1 | 5.2.1 | 5.2.1.4.1 | 5.2.1.4.1
- founder
- 5.2.1.1 | Construction | General
- FRET
- 5.2.2 | Discussion
- G-protein
- no title | 3.3 | 3.3 | Activity
- GAP
- 1.2.1 | 1.2.2
- GC-box
- 4.2
- GEF
- 1.2.2
- GPCR
- 1.2 | 3.3
- Grb2
- 1.2.1
- growth factors
- 1.2 | 1.2 | 2.2.1 | 2.2.1 | 2.3.1 | 2.3.3.1 | 2.4.2.1 | 2.4.2.7.1 | 3.5.2 | Context | Introduction | The
- haploinsufficient
- 2.4.3
- heat shock
- Abstract | 2.2.4 | 2.3.4.3.3 | 2.3.4.3.3 | 2.3.4.3.3 | Summary | Conclusion | Abstract | Discussion | Discussion
- HEK cells
- 2.4.2.1 | Cell | Discussion | Discussion
- HeLa cells
- 2.4.2.1 | 2.4.2.2 | 2.4.2.5 | 3.5.1 | Figures
- hinge
- 1.1.2 | 2.1.1 | 2.1.1 | 2.1.1 | 2.1.1 | 2.3.1.1 | 2.3.4 | 2.3.4 | 2.4.1 | 3.2
- histone
- ERK3 | 2.3.2.2 | 4. | 4. | 4.1 | 4.1 | 4.1 | 4.1 | 4.1
- hnRNP
- 2.3.3.1 | 2.3.4.3.6
- Hsp
- 2.3 | 2.3.4.1 | 2.3.4.3.3 | 2.3.4.3.3 | 2.3.4.3.4 | 2.3.4.3.4 | 2.4.2.4 | 2.4.2.5 | 2.4.2.6 | 2.4.2.6 | 4.1
- inflammation
- 1.1.2 | 2. | 2.3.4.3.3 | 5.1
- insulators
- 4.2
- kinome
- no title
- knockin
- 5.2 | 5.2.1.1 | 5. | Abstract | Background | Background
- knockout
- ERK3 | ERK4 | 2.3.1.2 | 2.3.3.2 | 2.3.4.3.3 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.7 | 2.3.4.3.7 | 2.3.4.3.7 | 2.3.4.3.7 | 2.4.2.7.3 | 2.4.3 | 2.4.3 | 2.4.3 | 3.4 | 5.2 | 5.2 | 5.2.1.4.2 | 5.2.1.4.3 | 5.2.1.6.1 | 5.2.2 | 5.2.2 | Summary | 3. | 4.1 | 4.2 | 1. | 1. | Background | Background | Anxiety-related | Anxiety-related | Anxiety-related | Activity | Activity
- LD
- Abstract | List | 5. | 1. | Background | Behavioural | Behavioural | Differences | Differences | Differences | Light-dark | Difference | Difference | Anxiety-related | Anxiety-related | Activity
- LErafAON
- 1.1.2
- LPS
- 2.2.4 | 2.2.4 | 2.3.4.3.3 | 2.3.4.3.3 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.7 | 2.4.3 | Materials | The | Discussion | Discussion | Figures | Figures
- MAP2K
- 1.1.1 | 2. | 2. | 2.
- MAP3K
- 1.1.1 | 1.1.1 | 2. | 2. | 2.
- MAPK
- 1.1.1 | 2. | 2. | 2. | 2.
- Mediator
- 4. | 4. | 4.3 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3
- microinjection
- 5.1 | Construction | Statistical | Statistical | Acknowledgements
- MIP2
- 2.3.4.3.6
- mitogens
- 2. | 2.
- MK2
- Abstract | 1.1.2 | 2.1.1 | 2.1.1 | 2.1.1 | 2.3 | 2.3 | 2.3 | 2.3.4 | 2.3.4.1 | 2.3.4.1 | 2.3.4.1 | 2.3.4.1 | 2.3.4.2.1 | 2.3.4.2.1 | 2.3.4.2.1 | 2.3.4.2.2 | 2.3.4.2.2 | 2.3.4.2.2 | 2.3.4.2.2 | 2.3.4.3.1 | 2.3.4.3.2 | 2.3.4.3.3 | 2.3.4.3.3 | 2.3.4.3.3 | 2.3.4.3.3 | 2.3.4.3.4 | 2.3.4.3.5 | 2.3.4.3.5 | 2.3.4.3.5 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.7 | 2.3.4.3.7 | 2.3.4.3.7 | 2.4 | 2.4.1 | 2.4.1 | 2.4.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.2 | 2.4.2.2 | 2.4.3 | 2.4.3 | 2.4.3 | Summary | 4.1 | Figures | Figures | Background | Background | Background | Abstract | Introduction | Introduction | Discussion
- MK3
- Abstract | 2.3 | 2.3 | 2.3 | 2.3.4 | 2.3.4.1 | 2.3.4.2.1 | 2.4.1 | 2.4.1 | 2.4.1 | Background | Background | Background
- MK4
- 2.3.4 | 2.4.1
- MKP
- List | 2.1.2 | 2.2.3 | 2.3.2.2
- NES
- 2.1.1 | ERK3 | 2.3.3 | 2.3.4 | 2.3.4 | 2.3.4.2.1 | 2.3.4.2.2 | 2.3.4.2.2 | 2.3.4.2.2 | 2.4.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.1 | 6 | 4.3 | 4.3 | 5. | 5. | 1. | Figures
- NG108-15 cells
- 3.5.2
- NIH3T3 cells
- 3.5 | 3.5.1 | Introduction
- NLS
- 2.1.1 | 2.3.3 | 2.3.4 | 2.3.4 | 2.3.4.2.1 | 2.3.4.2.1 | 2.3.4.2.2 | 2.3.4.2.2 | 2.4.1 | 2.4.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.1 | 6 | Figures
- NMDA
- 3.5.1
- octamer
- 4.2
- oncogene
- 1.2.2
- p38
- 1.1.1 | 2.1.1 | 2.1.2 | 2.2.2 | 2.2.4 | ERK3 | 2.3.2 | 2.3.2.2 | 2.3.3 | 2.3.4 | 2.3.4.1 | 2.3.4.1 | 2.3.4.2.1 | 2.3.4.2.1 | 2.3.4.2.2 | 2.3.4.2.2 | 2.3.4.2.2 | 2.3.4.3.1 | 2.3.4.3.2 | 2.3.4.3.3 | 2.3.4.3.4 | 2.3.4.3.5 | 2.3.4.3.7 | 2.4 | 2.4.1 | 2.4.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.1 | 2.4.2.2 | 2.4.2.7.1 | 2.4.2.7.2 | 2.4.3 | 3.5.1 | 4.1 | 5.2.1.6 | 3. | 4.3 | Anxiety-related | Introduction | The
- p53
- 2.3.4.3.5 | 2.3.4.3.5 | 2.4.2.4 | 2.4.2.4 | 3. | 3.
- PC12 cells
- 2.3.1.2 | Summary | 1. | 4.1 | 4.3 | Conclusion | 1. | Current | Current | Current | Figures | Figures | Figures | Construction | Abstract | Introduction | Introduction | Cell | Small | MK5 | MK5 | The | Discussion | Discussion | Discussion | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures
- PcG
- 2.3.4.1 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.7
- PDK1
- 2.3.1.1 | 2.3.2
- phosphatase
- List | 1.1 | 1.1.2 | 2.1.2 | 2.2.3 | 2.3.1.1 | 2.3.1.2 | 2.3.4.3.5 | 3.4 | 3.5.1 | Introduction | Materials
- phosphoacceptor
- 2. | 2.
- PI3K
- 1.2.2
- PKA
- Abstract | no title | 3.4 | 3.4 | 3.4 | 3.5 | 3.5.1 | 3.5.1 | 3.5.1 | 3.5.1 | 3.5.1 | 3.5.1 | 3.5.2 | 3.5.2 | 6 | Summary | 4.2 | 4.2 | 4.3 | Conclusion | 1. | Current | Current | MK5 | The | The | The | Discussion | Discussion | Figures | Figures | Figures | Figures | Figures | Figures | Figures
- PKI
- 2.4.1 | 2.4.1 | 3.3 | 1.
- promoter
- 3.3 | 3.5.1 | 4. | 4.1 | 4.2 | 4.2 | 4.2 | 4.2 | 4.2 | 4.2 | 4.2 | 5.1 | 5.2.1 | 5.2.1 | 5.2.1.2 | 5.2.1.4.1 | 5.2.1.5.1 | 5.2.1.6 | 5.2.1.6 | 5.2.1.6 | 6 | Summary | 1. | 1. | 1. | 4.2 | 1. | PCR | Statistical | Statistical | Abstract | Abstract | Plasmids | Plasmids | The | The | CREB | The | The | The | The | The | The | The | The | Discussion | Discussion | Discussion | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures | Figures
- pronucleus
- 5.1 | 5.2.1.1
- proto-oncogene
- 1.2.2
- Pyk
- 1.2.2
- pyrrolopyridine
- 1.1.2
- quantum dots
- 5.2.2 | 5.2.2 | 5.2.2
- Rac
- 1.2.2
- Raf
- 1.2.1 | 1.2.2 | 3.5 | 3.5.1 | 3.5.1 | 3.5.2 | Introduction
- Ran
- 1.2.2 | 1.2.2
- Ras
- 1.2.1 | 1.2.2 | 1.2.2 | 1.2.2 | 2.2.1 | 2.4.2.4 | 3. | Figures
- receptors
- 1. | 1.1.1 | 1.2 | 1.2.2 | 2.2.1 | 2.2.3 | ERK8 | 2.3.4.2.2 | 2.3.4.3.3 | 3.5.1
- Rho
- 1.2.2 | 1.2.2 | 2.2.3 | 4.1 | 4.1
- RNAi
- 1.1.2 | 5.2.1.4.3
- RNPII
- 4.2 | 4.2 | 4.2 | 4.2 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3
- SB203580
- 2.2.4 | 2.3.4.3.6 | 2.4.2.4 | 2.4.2.7.3
- scaffold
- 2.2 | 2.2.1 | 2.2.1 | 2.2.3 | 2.2.4 | 2.3.3.1 | 3.3 | 3.5.1
- senescence
- 2.4.2.4 | 2.4.2.4 | 2.4.2.4 | 2.4.3 | 2.4.3
- SH2
- 1.2 | 1.2.1 | 1.2.1
- SH3
- 1.2.1 | 2.1.1 | 2.2.2
- Shc
- 1.2 | 1.2.1
- shRNA
- 1.1.2 | 2.4.2.4 | 5.2.1.1 | 5.2.1.3 | 5.2.1.4.3
- siRNA
- 1.1.2 | Summary | 1. | Figures | Abstract | Small | The | Figures
- SK-N-DZ cells
- Cell | MK5 | The | Discussion | Figures | Figures | Figures | Figures | Figures | Figures
- Sorafenib
- 1.1.2
- SOS
- 1.2.1
- Src
- 1.2.1 | ERK8
- TATA box
- 4.2 | 4.2 | 4.2 | 4.2 | 4.2 | 4.3 | 1. | The | The
- TBP
- 4.2 | 4.3
- tetracycline
- 5.2 | 5.2.1.5.1
- TNF-alpha
- 2.3.3.2 | 2.3.4.3.3 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.7 | 2.4.2.1 | 2.4.2.5 | 2.4.3
- TTP
- 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.6 | 2.3.4.3.7 | 2.3.4.3.7
- viral vectors
- 5.2.1.3
About this document ...
This document was generated using the LaTeX2HTML translator Version 2002-2-1 (1.71)
Copyright © 1993, 1994, 1995, 1996, Nikos
Drakos, Computer Based Learning Unit, University of Leeds.
Copyright
© 1997, 1998, 1999, Ross
Moore, Mathematics Department, Macquarie University, Sydney.
The translation was initiated on 2007-12-08