





Publications
Presentations/Lectures
Modulation of F-actin rearrangements by the cyclic AMP/PKA pathway is mediated by MAPKAP Kinase 5 and requires PKA-induced nuclear export of MK5
Nancy Gerits*, Theresa Mikalsen*, Sergiy Kostenko, Alexey Shiryaev, Mona Johannessen and Ugo Moens#
Department of Microbiology and Virology, Faculty of Medicine, University of Tromsø, N-9037 Tromsø, Norway.
#Corresponding author: Ugo Moens, Department of Microbiology and Virology, Faculty of Medicine, University of Tromsø, N-9037 Tromsø, Norway, Phone:+47-776 44622, Fax: +47-776 45350, e-mail: ugom@fagmed.uit.no. * Both authors contributed equally. ``Journal of Biological Chemistry'' October 2007. In Press.
Abstract
The mitogen-activated protein kinase (MAPK)-activated protein kinases belong to the Ca2+/calmodulin-dependent
protein kinases. Within this group, MK2, MK3 and MK5 constitute three structurally related enzymes with distinct functions. Few genuine substrates for MK5 have been identified and the only known biological role is in ras-induced senescence and in tumour suppression. Here, we demonstrate that activation of PKA or ectopic expression of the catalytic subunit Cα in PC12 cells results in transient nuclear export of MK5, which requires the kinase activity of both Cα and MK5 and the ability of Cα to enter the nucleus. Cα and MK5, but not MK2, interact in vivo and Cα increased the kinase activity of MK5. Moreover, Cα augments MK5 phosphorylation, but not MK2, while MK5 does not seem to phosphorylate Cα. Activation of PKA can induce actin filament accumulation at the plasma membrane and formation of actin-based filopodia. We demonstrate that siRNA-triggered depletion of MK5 interferes with PKA-induced F-actin rearrangements. Moreover, cytoplasmic expression of an activated MK5 variant is sufficient to mimic PKA provoked F-actin remodelling. Our results describe a novel interaction between the PKA pathway and MAPK signalling cascades, and suggest that MK5, but not MK2 is implicated in PKA-induced microfilament rearrangements.
Introduction
Mitogen activated protein (MAP)kinase signalling
pathways play important roles in several cellular processes,
including proliferation, differentiation, apoptosis, development,
gene transcription, and motility. A typical MAP kinase module acts
through subsequent phosphorylation and activation of a MAP kinase
kinase kinase, a MAP kinase kinase, and a MAP kinase. The MAP kinase
may phosphorylate non-protein kinase substrates or yet other protein
kinases, referred to as MAPK-activating protein kinases (MAPKAPK)
[1-5]. Based on the sequence homology of
the kinase domain, the MAPKAPK belong to the superfamily of
Ca2+/calmodulin-dependent protein kinases [6].
The mammalian MAPK APK include the ribosomal-S6-kinases (RSK1-4 or
MAPKAPK1a-d), the mitogen-and stress-activated kinases (MSK1 and 2),
the MAPK-interacting kinases (MNK1 and 2) and the MAPKAPK MK2, MK3
and MK5. MK2 was isolated 15 years ago in an attempt to identify
protein kinases involved in adrenergic control of glycogen synthase
activity, while MK3 was first described as a protein encoded by a
gene located in the small cell lung cancer tumour suppressor gene
region 3p21.3 [7-8]. The murine MAPKAPK5
(MK5) or its human homologue PRAK was originally described as a
downstream target of p38 MAPK [9-10].
MK5 shares approximately 45% sequence homology with MK2 and MK3,
while MK2 and MK3 display 75% sequence identity. All three kinases
contain the same conserved regulatory phosphorylation site (Thr182,
Thr222, Thr201, respectively). Despite their high similarity, gene
deletion experiments reveal that these three kinases possess separate
functions [11-12]. MK2 is involved
in many cellular processes like cell cycle regulation, inflammation
and cell migration [13-18], whereas
the biological roles of MK3 and MK5 remain unclear. MK3 may be
involved in transcription regulation through modulating the activity
of the transcription factor E47 and the transcriptional repressor
Polycomb group Bmi-1 protein. Moreover, MK3 may function as a tumour
suppressor [19-21]. The biological
function of MK5 is poorly understood, as MK5 knockout mice bred onto
different backgrounds displayed either no obvious phenotype or
embryonic lethality. The reason for this lethality, however, remains
unknown. Furthermore, the atypical MAP kinases ERK3 and ERK4 are the
only identified in vivo MK5-interacting partners, but the
physiological relevance of the interaction remains
elusive [11,22-26]. However, a
recent study demonstrated that MK5 could phosphorylate p53 at serine
residue 37 in vivo, that MK5 deficiency enhanced mutagen-induced skin
carcinogenesis in mice, and that MK5-/- primary cells were
more susceptibility to oncogenic transformation. These findings point
to a role for MK5 in tumour suppression [27].
Our group, in collaboration with others, found that increased levels
of ERK3 during nerve growth factor-induced differentiation of PC12
cells was accompanied by increased MK5 activity, hence suggesting a
role for MK5 in neuronal differentiation [24].
Cyclic
AMP (cAMP) functions as an intracellular second messenger to transmit
extracellular signals, such as hormones and neurotransmitters, to the
intracellular environment. The major target of cAMP is the
cAMP-dependent protein kinase (PKA), a tetrameric holoenzyme that
consists of two regulatory (R) and two catalytic (C) subunits. After
cAMP binds the R subunits, the C subunits dissociate and
phosphorylate their substrates [28]. The
cAMP/PKA signalling pathway regulates a variety of cellular processes
such as cell proliferation, differentiation, actin cytoskeletal
rearrangements, and gene transcription (reviewed in [29-31]).
In order to optimise the responsiveness to a wide variety of signals,
the PKA pathway interacts with other signalling pathways, including
the MAPK pathways. In fact, PKA has been shown to modulate the
activity of several MAPK cascades [32-35].
Recently, the MAPKAP kinase, RSK1, was found to either bind the
catalytic C or regulatory R subunit of PKA, depending on the
phosphorylation status of RSK1, but the physiological relevance of
this interaction is not completely understood [36].
A role for PKA in actin cytoskeletal changes is well
illustrated, and it has been suggested that MK2/3/5 may be implicated
in actin remodelling (reviewed in [11]
and [13]). However, whether there is a
specific role for a certain MK in actin redistribution and whether
PKA may recruit MKs to mediate actin rearrangement remains unsolved.
This prompted us to examine whether MK5 might contribute in
cAMP/PKA-provoked modification of the microfilament architecture in
the rat pheochromocytoma cell line PC12. Our results demonstrate a
new interaction between the cAMP/PKA and MK5, and recognise a novel
role of MK5 as a mediator of cAMP-induced F-actin rearrangements.
Experimental Procedures
Materials
Forskolin, PKA-C and PKA-R were purchased from Sigma Aldrich (St. Louis, MO, USA). 32P δ-ATP was obtained from Amersham/GE Healthcare (Buckinghamshire, UK), while recombinant active and inactive His-MK5/PRAK were from Invitrogen (Carlsbad, CA, USA). The PRAKtide MK5 substrate peptide was purchased from Upstate (Charlottesville, VA, USA). The monoclonal antibody against the regulatory RIα of PKA was a kind gift from Dr. Skålhegg (University of Oslo). Anti-PRAK (A-7), anti-ERK2 (C-14) and anti-PKAcat (C-20) were all from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). Anti-phospho-CREB (#9190) and anti-MK2 (#3042) were obtained from Cell signalling (Beverly, MA), while anti-GFP (ab290) was purchased from AbCam (Cambridge, UK). Anti-ERK3 antibodies have been described before [24]. The monoclonal anti-Ras clone RAS10 antibody was from Upstate (cat. no. 05-516; Charlottesville, VA, USA). Alexa Fluor 488 antibody was from Invitrogen. The alkalic phosphatase-conjugated secondary antibodies sheep anti-mouse IgG and anti-rabbit IgG were from Sigma Aldrich. Oligonucleotides were purchased from Sigma Aldrich or Eurogentec (Seraing, Belgium).
Plasmids
The expression vectors for p38β
and the activated MKK6E/E mutant, as well as for the EGFP fusion
proteins with wild-type MK5, and the MK5 mutants K51E, T182A and
L337A, and truncated MK5 1-423 have been previously described
[24,37]. Dr. Maurer generously provided
the expression plasmid of the heat-stable inhibitor of PKA (pCMVPKI)
[38]. The Cα-NLS
plasmid was a kind gift of Dr. Thiel [39].
The kinase dead Cα
L72H was generated by site-directed mutagenesis using the primer
(only one strand is shown):
5'-GAA-CAA-CTA-CGC-CAT-GCA-CAT-CTT-AGA-CAA-G-3'. The expression
plasmids for cytoplasmic located MK5 variants were generated by
cloning the NES signal of the Rev protein of human immunodeficiency
virus into
pEGFP-MK5. Tagging a protein with this sequence will
direct it to the cytoplasm [40].
Thereto, the complementary oligonucleotides
5'-CCGGAGACGCTCTACCACCGCTTGAGAGACTTACTCTTGACCGAGCT-3' and
5'-CGGTCAAGAGTAAGTCTCTCAAGCGGTGGTA GAGCGTCT-3' were ligated into the
BspEI/Sac I sites of pEGFP-MK5, pEGFP-MK5 T182A, and pEGFP-MK5 L337A,
respectively. To construct the RFP-Cα-NLS
and RFP-Cα-NES
expression plasmids, the BspEI and BamHI fragment of GPKAnls,
respectively GPKAnes, was ligated into the corresponding sites of
pAsRed2-C1 (Clontech, Mountain View, CA, USA). The GPKAnls and
GPKAnes plasmids were
generously provided by Dr. S.H. Green [41].
The plasmid pCRE-LUC was from Clontech. All newly generated plasmids
and mutations were confirmed by sequencing.
In vitro kinase assay
Phosphorylation of purified MK2 (Upstate Millipore, Billerica, MA, USA), MK5 (Upstate Millipore), and ERK3 [24] by 2.5 units PKA-C was performed in 25 mM Tris.HCl pH7.5, 10 mM MgCl2, 0.05 mg/ml BSA, 2.5 mM DTT, 0.15 mM cold ATP and 0.3 μl 32P γ-ATP(3000 Ci/mmol; Amersham/GE Healthcare) in a total volume of 40 μl at 30°C for 1 hour. The reaction was stopped in 4xLDS Sample buffer and proteins denatured at 70°C for 10 minutes. The phosphorylation was analysed on NuPage 4-12% Bis-Tris SDS-PAGE (Invitrogen) for 50 minutes at 200V, then subjected to autoradiography.
Cell culture and transfection
PC12 cells, a kind gift from Dr. Jaakko Saraste
(University of Bergen, Norway), and PKA-deficient PC12A123.7
cells (generously provide by Dr. J. Yao) were maintained in
RPMI-1640, supplemented with 10% horse serum (Gibco) and 5% foetal
bovine serum, 2 mM L-glutamine, penicillin (110 units/ml) and
streptomycin (100 μg/ml).
HeLa cells were maintained in Eagle's Minimum Essential Medium
supplemented with 1x non-essential amino acids, 10% foetal calf
serum, 2 mM L-glutamine, penicillin (110 units/ml) and streptomycin
(100 μg/ml). HEK293
cells were purchased from the European Collection of Cell cultures
(cat. no. 85120602; Salisbury, Wiltshire, UK) and kept in Eagle's
Minimum Essential Medium supplemented with 10% foetal calf serum, 2
mM L-glutamine, penicillin (110 units/ml) and streptomycin (100
μg/ml). Cells were
transfected with Lipofectamine 2000 (Invitrogen) or using the
Nucleofection kit (Amaxa) according to the manufacturers
instructions. We routinely obtained >75% transfection efficiency
in the different cell lines with these transfection agents as judged
by green fluorescent positive cells when using the pEGFP plasmid as a
marker.
Preparation of nuclear and cytoplasmic extracts
The NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL, USA; cat. no. 78833) was used according to the instructions of the manufacturer.
Western blotting
For detection of co-immunoprecipitates and MK5, samples were analysed by SDS-PAGE NuPage 4-12% Bis-Tris SDS-PAGE (Invitrogen) according to the manufacturers protocol and blotted onto a 0.45 μm PVDF membrane (Millipore, Billerica, MA, USA). Immunoblotting was performed by first blocking the membrane with PBS-T (PBS with 0.1% Tween-20 (Sigma Aldrich) containing 10% (w/v) dried skimmed milk for 1 hour and probed either by 4D7, anti-PRAK, anti-MK2, anti-ERK2 or anti-PKA cat. After 4 washes, the membrane was incubated with the appropriate secondary antibody for 1 hour. Visualisation of proteins was achieved by using CDP Star (Tropix, Bedford, MA, USA) substrate and Lumi-Imager F1 from Roche (Basel, Switzerland).
Immunoprecipitation
HeLa extracts were harvested and lysed in buffer containing 20 mM Tris.HCl pH 7.5, 1% Triton X-100, 5 mM sodium pyrophosphate, 50 mM sodium fluoride, 1 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate, 0.27 M sucrose, 10 mM β-glycerophosphate, and Complete Protease Inhibitor Cocktail (Roche). Lysates were cleared by centrifugation at 4°C for 10 minutes at 15,000g. Lysates were incubated with the appropriate antibody for at least 1 hour at 4°C, before addition of 60 μl slurry (i.e. 30 μl protein G-agarose (Amersham/GE Healthcare) equilibrated with 30 μl lysis buffer) and incubated for an additional hour. The immunoprecipitates were then washed three times in lysis buffer and twice in 50 mM Tris-Cl pH8.0. Twenty μl 2xLDS sample buffer was added to the beads before denaturation at 70°C for 10 minutes. The immunoprecipitates were either analysed on Western blots or by means of kinase assays.
Cell staining and microscopy
Cells were rinsed twice with phosphate-buffered saline (PBS) and fixed for 10 min with 4% formaldehyde. Next, the cells were washed twice with PBS and then permeabilised for 10 min with 0.1% Triton X-100. The cells were pre-incubated with PBS containing 1% BSA for 20-30 min and stained with Alexa Fluor 594 phalloidin (#A12381; Molecular Probes, Eugene, OR) for 20 min. The cells were then washed with PBS and examined using confocal laser-scanning Zeiss LSM 510 META and Leica SP5 microscopes. Cell nuclei were visualised by DRAQ5 staining (Biostatus Limited, Leicestershire, United Kingdom). We routinely examined 50 cells or more and representative pictures are shown in the results.
Transient transfection and luciferase assay
HEK293 cells were plated 3x105 cells per well in a 6-wells plate. Cells were transfected using Lipofectamine 2000 according to the manufacturer's instruction. Luciferase assays were as previously described [42].
Immune complex kinase assay
For the immune complex kinase assays, HeLa cells
were transfected with expression plasmids encoding EGFP fusion
proteins with wild-type MK5 or mutants and Cα.
Corresponding empty vector control plasmids (pEGFP and pRcCMV,
respectively) were used as control. As a positive control for the
assay, cells were either transfected with pEGFP-MK5L337A,
encoding an activated MK5 variant, or pEGFP-MK5 plus expression
plasmids for the activated MKK6E/E mutant and p38β
MAPK. MK5 fusion proteins were immunoprecipitated using anti-GFP
antibody and the kinase activity was monitored against 30 mu M
PRAKtide substrate peptide at 30°C for 20 minutes in 50 mu l
buffer containing 40 mM TrisCl pH 7.5, 0.4 mg/ml BSA, 15 mM MgCl2,
0.1 mM cold ATP, and 1 μCi
32P gamma
-ATP (3000Ci/mmol). Following incubation, 20
mu l aliquots were spotted onto phosphocellulose discs (Upstate) and
washed extensively with 1% phosphoric acid before measurement of
radioactive incorporation by scintillation counting.
Quantitation of F-actin
The amount of globular (G-actin) and filamentous actin (F-actin) was determined using the G-actin/F-actin In Vivo Assay Kit from Cytoskeleton (Denver, CO, USA) according to the instructions of the manufacturer. Briefly, cells were lysed and the lysate was centrifugated at 100,000g for 1h at 37°C. The supernatant contained G-actin, while the pellet contained F-actin. As a control, cell lysates were either treated with F-actin enhancing solution (phalloidin) or F-actin polymerisation solution (cytochalasin) before ultracentrifugation. The amount of G- and F-actin was determined by western blotting using an anti-actin specific antibody.
Small interfering RNA (siRNA)
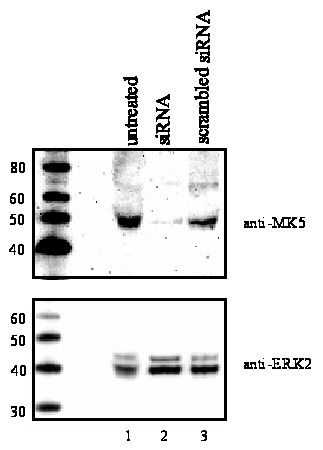
Validated MK5-directed siRNA, purchased from Ambion Inc. (Austin, TX), was transfected by Nucleofection (Amaxa) into PC12 cells according to the manufacturers instructions using 100 mu M siRNA/106cells. The levels of MK5 protein in untreated and siRNA treated cells were monitored 48 hours after transfection by western blotting using anti-PRAK antibody (Santa Cruz) to verify the efficiency of reduction in MK5 expression.
Results
The cAMP/PKA pathway induces nuclear export of MK5
Both endogenous and ectopically expressed MK5 as
green fluorescent protein (EGFP)-, cyan fluorescent protein-, and
HA-tagged proteins reside predominantly in the nucleus in different
cells [23, 37, 43]. Studies have proven
that the cAMP/PKA signalling pathway participates in actin
remodelling, and that MKs may regulate the cytoskeletal architecture
through modulating the activity of cytoplasmic proteins involved in
F-actin organisation (reviewed in [11]
and [30]). Because the putative
involvement of MK5 in stimulus-induced microfilament rearrangement
raises the likelihood of cytoplasmic distribution of MK5, we tested
whether activation of PKA triggered subcellular redistribution of MK5
in PC12 cells. Thereto cells transfected with an expression plasmid
for EGFP-MK5 fusion protein were treated with the cAMP-elevating
agent forskolin. Consistent with previous findings of both ectopic
and endogenous MK5, we observed MK5 mainly in the nucleus of
unstimulated cells [23, 37, 43]. A
transient nuclear export of MK5 was observed in cells treated with
forskolin, but not in control cells treated with the vehicle DMSO.
Increased cytoplasmic MK5 residence was observed 15 min after
forskolin exposure and maintained elevated for at least 1 hour, after
which EGFP-MK5 relocalised mainly to the nucleus. After 30 minutes of
forskolin stimulation, the amount of cells displaying both nuclear
and cytoplasmic EGFP-MK5 versus nuclear EGFP-MK5 alone increased from
6% to 84% (50 cells were counted). After 6 hours, the subcellular
distribution of MK5 was comparable to untreated cells (Figure
1A). In 95% (+/- 2%; 50 cells were counted in several
independent experiments) of the DMSO-treated cells, MK5 was
predominantly nuclear. Forskolin treatment had no effect on the
subcellular localisation of the EGFP protein (results not shown).
Endogenous MK5 was also exported from the nucleus in cells with the
PKA signalling pathway activated with the same kinetics as EGFP-MK5
(Figure 1B). These findings confirm that
MK5 and EGFP-MK5 fusion protein behave identically in their
subcellular redistribution in response to forskolin. Monitoring the
amount of MK5 in the cytoplasmic and nuclear fraction of untreated
and forskolin-treated cells confirmed an increased nuclear export of
MK5 after forskolin exposure. A 55% decrease of nuclear MK5 was
observed after 30 min forskolin treatment, while a 41% increase in
cytoplasmic MK5 was monitored in forskolin-exposed cells (Figure
1C).
Next, we tested whether overexpression of the
catalytic subunit of PKA tagged with a nuclear localisation signal
(Calpha -NLS) was sufficient to induce nuclear export of MK5. Similar
to forskolin-exposed cells, MK5 was located in the cytoplasm of cells
that ectopically expressed Calpha -NLS (Supplementary
data, Figure 1). To elaborate the contribution of PKA in
forskolin-induced nuclear export of MK5, we used PC12A123.7
cells, which are deficient in PKA [44].
No subcellular redistribution of MK5 was monitored in these cells
after forskolin treatment (Supplementary data,
Figure 2). In conclusion, these results demonstrate that
activation of the cAMP/PKA signalling pathway provokes nuclear export
of MK5 in a time-dependent manner.
The catalytic Cα subunit of PKA phosphorylates MK5 and increases the kinase activity of MK5
Previous studies by our group and others have shown that nuclear export of MK5 can be achieved by overexpression of the atypical MAP kinases ERK3 and ERK4. This nuclear exclusion requires the direct interaction between MK5 and ERK3 or ERK4, and leads to activation of MK5 [23-26]. To find out whether PKA can interact with MK5, HeLa cells were transfected with EGFP-MK5 and Calpha expression plasmids and reciprocal co-immunoprecipitation studies were performed with anti-Calpha antibody followed by western blotting with MK5 antibody, or co-immunoprecipitation with anti-EGFP antibody followed by western blotting with anti-Calpha antibody. A specific interaction was monitored between these two proteins (Figure 2A). The positive signal in Fig. 2A, lane 6 (lysates of cells ectopically expressing EGFP-MK5 but not Calpha ) is probably the result of interaction between ectopically expressed MK5 and endogenous Calpha . No interactions were detected in the control pull-down assays using empty expression vectors. Because of the sequence similarity between MK2 and MK5, we investigated whether Calpha also associates with MK2. Cells were co-transfected with EGFP-MK2 and Calpha expression vectors or empty vectors as controls and lysates were immunoprecipitated with anti-Calpha or anti-EGFP antibodies. The presence of the co-immunoprecipitated partner was detected by western blot using anti-MK2 or anti-Calpha antibodies, respectively. However, under these experimental conditions no interaction between MK2 and Calpha was detected (Figure 2B). The MAPKAP kinase RSK1 was reported to interact with both the Calpha and the regulatory subunit of PKA [36]. This prompted us to test whether such an interaction also occurred with MK5. No association between MK5 and the regulatory RIalpha subunit of PKA was observed (results not shown). Next, we examined whether PKA, in agreement with the MK5-interacting protein kinases ERK3 and ERK4, possessed the ability to phosphorylate MK5 and activate its intrinsic kinase activity. An in vitro kinase assay demonstrated that both Calpha and MK5 possessed autophosphorylation activity, and that incubation of purified Calpha with purified MK5 increased the phosphorylation levels of MK5 approximately 2.5-fold, but not of Cα (Figure 3A). Calpha did not phosphorylate MK2, but phosphorylated the myelin basic protein (MBP), which was used as a positive control (Figure 3B). The inability of Cα to phosphorylate MK2 is in concordance with our previous findings that MK2 and Calpha did not interact in vivo (Figure 2B). To monitor the effect of PKA on the enzymatic activity of MK5, we performed an in vitro kinase assay. ERK3 was selected as a substrate for MK5. ERK3 displayed low levels of autophosphorylation activity. Cα alone or inactive MK5 alone did not induce phosphorylation of ERK3. However, incubation of Cα plus inactive MK5 strongly increased the phosphorylation levels of ERK3 (Figure 4A, top panel). Western blotting with anti-ERK3 antibodies ensured that equal amounts of ERK3 substrate were used in the samples (Figure 4A, bottom panel). These results suggest that Calpha enhanced the kinase activity of MK5 towards ERK3. Subsequently, we tested the effect of PKA on the in vivo kinase activity of MK5 using an immune complex kinase assays. Thereto, a series of co-transfections with EGFP-MK5 and Cα-NLS expression plasmids or empty vectors (pEGFP-C1 and pRcCMV, respectively) were performed and the protein kinase activity of MK5 towards the synthetic PRAKtide substrate was measured. While EGFP alone or in the presence of Cα had no kinase activity towards PRAKtide (results not shown), EGFP-MK5 phosphorylated PRAKtide. Co-expression of Cα enhanced the kinase activity of MK5 approximately 2-fold in most experiments (n=6), although occasionally we did not observe an increase of MK5 activity in the presence of Cα (n=2). The constitutive active MK5 mutant L337A [37] and an activated MK5 by co-expression of active MKK6 E/E and p38 MAPK possessed a 3- to 5-fold increased kinase activity compared to wild-type MK5 (Figure 4B). Forskolin stimulation alone also induced the kinase activity of MK5 (results not shown). In conclusion, Cα physically interacts with and stimulates the kinase activity of MK5 in vivo.
PKA-induced nuclear export of MK5 requires nuclear localisation of Cα
The co-immunoprecipitation studies suggest an in
vivo interaction between Cα
and MK5, but they do not establish whether Cα
is directly involved in the subcellular redistribution of MK5. We
therefore wanted to determine whether nuclear entrance of Cα
is required to induce nuclear export of MK5. We monitored the
subcellular localisation of MK5 in forskolin-treated cells that
overexpress PKI, a peptide that contains a strong nuclear export
signal and that can export Calpha out of the nucleus (for a recent
review see [45]).
Overexpression of
PKI prevented forskolin-induced nuclear export of MK5 in PC12 cells.
Of the EGFP-MK5 expressing cells, in 98% of the cells (+/- 2%; 50
cells were counted in several independent experiments) MK5 was mainly
in the nucleus (Figure 5A). This is in
contrast with the transient nuclear exclusion of MK5 in
forskolin-treated PC12 cells (Figure 1).
To further ascertain the necessity of nuclear entry of Cα
in MK5 cytoplasmic relocation, we compared the residence of EGFP-MK5
in cells co-transfected with an expression plasmid for EGFP-MK5 plus
an expression vector for Cα
tagged with either a nuclear localisation signal (NLS) or with a
nuclear export signal (NES). In order to visualise Cα
and MK5 simultaneously, we used red fluorescence protein-Calpha
fusion proteins (AsRed-PKA-NLS and AsRedPKA-NES, respectively). The
NES-tagged Cα
subunit resided in the cytoplasm and was unable to induce nuclear
export of MK5, while in cells expressing a Cα-NLS
fusion protein both cytoplasmic and nuclear MK5 location was observed
(in 90% +/- 2% of the analysed cells; Figures
5B and 5C). The levels of cytoplasmic MK5 in Cα-NLS
expressing cells were increased compared to cells not expressing
Calpha -NLS (compare untreated cells in Figures
1A and 5C).
PKA-induced nuclear export of MK5 requires protein kinase activity of PKA as well as MK5
Next, we investigated whether the kinase activity of
Calpha was required to provoke nuclear export of MK5. Thereto,
expression plasmids for the kinase dead Cα
mutant (CL72H) and EGFP-MK5 were co-transfected in PC12
cells and subcellular localisation of MK5 was monitored. In contrast
to an active Cα
enzyme, no nuclear export was observed with the mutant catalytic PKA
subunit. In fact, in 98% (+/- 2%; 50 cells were counted in several
independent experiments) of the cells, MK5 resided predominantly
in the nucleus (Figure 6A). Western blot
analysis of cell extracts transfected with the kinase dead Cα
L72H mutant confirmed that this mutant had no enzymatic activity
because phosphorylation of the transcription factor CREB, which is a
genuine substrate for PKA, did not increase in the presence of this
mutant, while increased phospho-CREB was detected in
extracts
prepared from cells transfected with the active Cα
expression plasmid. Moreover, the kinase dead Cα
mutant was unable to stimulate CRE-dependent transcription (bottom
panel Figure 6A). To decide whether MK5
enzyme activity is required for PKA-regulated nuclear export, we
monitored the subcellular distribution of the kinase dead MK5 K51E
mutant and the T182A mutation in the phosphorylation loop. Both
mutants also failed to relocate to the cytoplasm in the presence of
activated PKA (Figure 6B). In agreement
with our observations in PC12 cells, forskolin treatment of HeLa
cells also induced transient nuclear export of EGFP-MK5 protein, but
not of the kinase dead T182A mutant, indicating a non cell-specific
mechanism (results not shown). These findings indicate that both an
active Cα and active
MK5 are required for PKA-induced cytoplasmic translocation in PC12
cells.
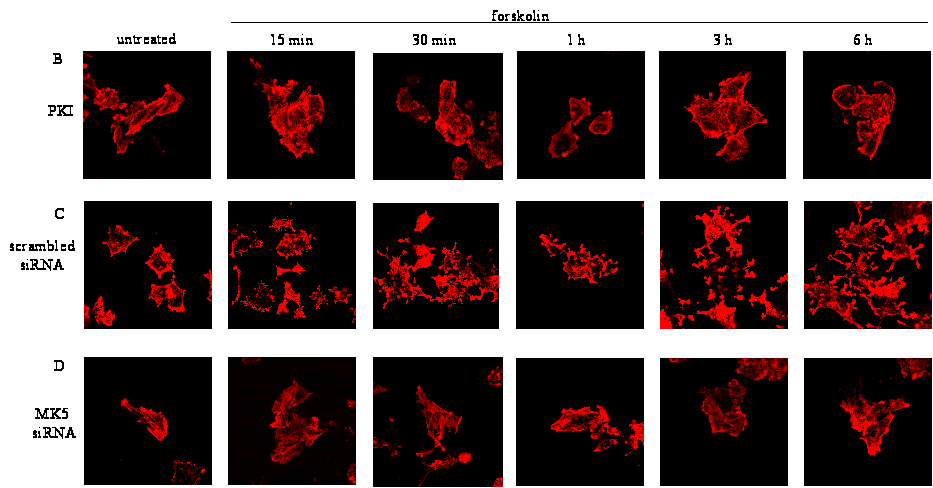
MK5 mediates forskolin-induced F-actin rearrangements
Others and we noticed that forskolin treatment of
PC12 cells resulted in altered cytoskeleton morphology [11,30].
Therefore, we assessed the amount of F-actin in response to
forskolin. A transient increase in F-actin was observed 30 min after
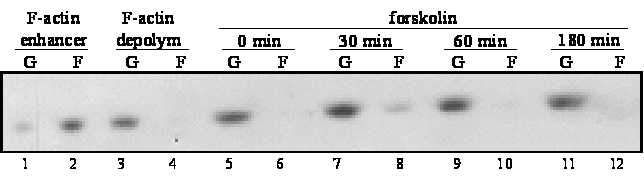
forskolin treatment (Figure 7A).
Interestingly, the kinetics of F-actin rearrangement and increase
coincided with the nuclear exclusion of MK5, presuming the
involvement of the MK5 in PKA-induced F-actin remodelling. To address
the possible implication of MK5 in cAMP/PKA-induced F-actin
rearrangement, several different experimental approaches were
conducted. We reasoned that the coupled event nuclear import of
Cα/cytoplasmic
export of MK5 would be required for F-actin reorganisation.
Therefore, we compared the microfilament structure in untreated and
forskolin-treated non-transfected cells or cells transfected with a
PKI expression plasmid. F-actin remodelling was inhibited in cells
overexpressing PKI (>75% of the PKI transfected cells did not
display F-actin rearrangement after forskolin exposure; Figure
7B). This underscores that nuclear import of PKA is necessary
and suggests that nuclear export of MK5 may be required for
cAMP/PKA-induced F-actin remodelling. Next, we examined
forskolin-induced F-actin rearrangement in control cells and in cells
in which the levels of MK5 protein had been depleted by validated
siRNA against MK5. In agreement with previous reports, we observed
that forskolin treatment of PC12 cells, which had been transfected
with scrambled siRNA, resulted in F-actin remodelling (Figure
7C). However, the majority of cells (>75%; 50 cells counted
in two independent experiments)
treated with MK5-directed siRNA
did not show F-actin rearrangements after forskolin treatment (Figure
7D). While siRNA directed against MK5 transcripts strongly
reduced MK5, but not ERK2 protein levels, scrambled siRNA did not
reduce the levels of either of these proteins (Figure
7E). Taken together, these results indicate that MK5 mediates
forskolin-induced F-actin remodelling. To elaborate the role of
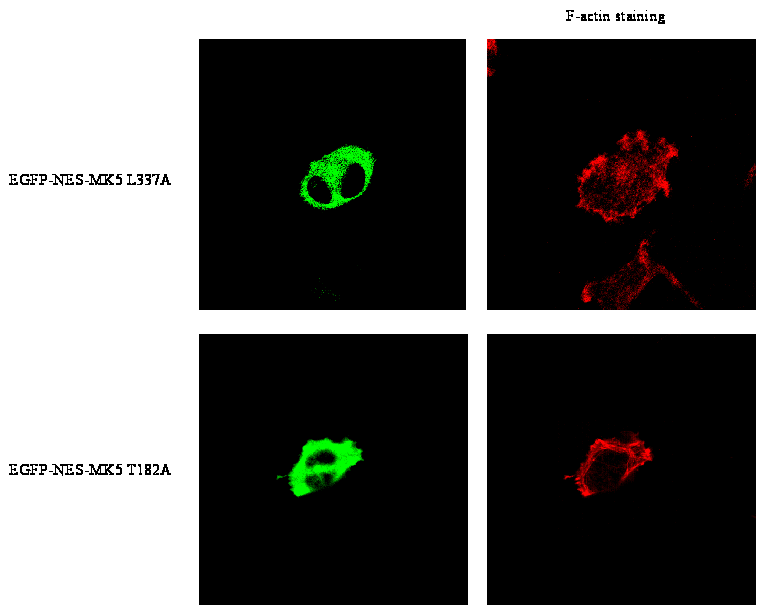
activated, cytoplasmic MK5 in F-actin rearrangement, we generated a
MK5 mutant that is constitutive active and that locates to the
cytoplasm. Our previous study had shown that a single amino-acid
substitution in the functional NES motif of MK5 (L337A) converted MK5
in a constitutive active kinase. However, this mutant has lost its
ability to shuttle to the cytoplasm [37].
To conduct this active MK5 variant to the cytoplasm, we fused the NES
sequence of the human immunodeficiency virus (HIV) Rev protein to
EGFP-MK5 to generate the fusion protein EGFP-NES-MK5 L337A. Studies
by others had shown that this NES motif
could direct nuclear
exclusion of EGFP [40]. Concordantly,
the EGFP-NES-MK5 L337A protein resides exclusively in the cytoplasm
(Figure 7F, top panel). All cells
expressing this cytoplasmic, activated MK5 mutant (50 cells counted
in three independent experiments) could induce some F-actin
rearrangement in the absence of forskolin (Figure
7F, top panel). Overlay of the confocal microscopy images
confirmed F-actin rearrangements in cells transfected with this MK5
mutant (results not shown), while no such changes were observed in
non-transfected cells. Cytoplasmic expression of EGFP-NES-MK5 T182A
(Figure 7D bottom panel) or EGFP-NES-MK5
(results not shown) did not affect the microfilament structure in
these cells, despite the cytoplasmic location of MK5. These results
proof that an activated, cytoplasmic MK5 can partially mimic
forskolin-induced F-actin rearrangement, and suggest that
PKA-dependent nuclear export and activation of MK5 is implicated in
PKA-triggered reorganisation of F-actin.
Discussion
The MAPKAPK MK5 was first described almost 10 years ago, but as to date little is known about its activators, in vivo substrates, and biological functions. The atypical MAPKs ERK3 and ERK4 were the first identified genuine substrates for MK5, but the functional importance of these interactions remains unclear [23-26]. The original study with MK5-/- mice revealed no obvious phenotypical defects [22], but a recent study with MK5 deficient mice led to the recognition of the implication of MK5 in ras-induced senescence and tumour suppression in a p38 MAPK-dependent manner through activation of p53 [27].
Results from our studies assign
-
a novel mode of regulating the subcellular localisation of MK5,
-
PKA as a new activator for MK5, and
-
a not previously recognised biological function of MK5 in F-actin rearrangements.
MK5, which contains functional, but overlapping NLS
and NES motifs, accumulates in the nucleus of resting cells. The NLS
signal seems to be dominant over NES, probably due to the
conformation of the MK5 protein in which the
NES is less
accessible to exportin. However, in response to cellular stress, the
NES motif may be unmasked and MK5 can be exported to the cytoplasm
[37,43]. In this study, we observed that
activated PKA triggered nuclear export of MK5. Time studies revealed
that at least 15 min were required for MK5 to exit the nucleus in
response to forskolin (Figure 1), while
nuclear translocation of the catalytic subunit of PKA in response to
forskolin was reported to plateau after 15 minutes [46].
This similar kinetics may indicate that nuclear import of activated
PKA is necessary for nuclear export of MK5. Cytoplasmic
redistribution of MK5 is also observed when ERK3 and ERK4 are
overexpressed. The mechanisms of ERK3/ERK4-mediated nuclear export of
MK5 differ, however, from Calpha -mediated export. Co-expression of a
kinase dead Calpha did not lead to accumulation of MK5 in the
cytoplasm, and similarly, the kinase dead MK5 variants K51E and T182A
were not excluded from the nucleus by an active Cα
or forskolin.
On the contrary, kinase dead ERK3 and ERK4 mutants
were able to induce nuclear export of wild-type MK5, but also of
catalytic dead MK5 mutant proteins [23-26].
Further studies that identify the interaction regions on MK5 will
provide additional information on the mechanistic insight how Calpha
regulates the nucleocytoplasmic relocation of MK5. Our preliminary
results show that a C-terminal truncated MK5 consisting of residues
1-423 is not transported out of the nucleus upon forskolin treatment.
On the other hand, ERK3 still relocates this truncated MK5 mutant to
the cytoplasm [24], indicating that
nuclear exclusion of MK5 by PKA is not mediated by ERK3. ERK4,
however, does not translocate this MK5 variant to the cytoplasm.
Whether ERK4 is implicated in PKA-induced nuclear export of MK5
remains to be established. The C-terminal part of MK5, which is
absent in MK2 and MK3, may be critical for PKA-induced MK5 nuclear
export. Since the carboxy-terminal part of MK5 is absent in MK2 and
MK3, it may explain the unique role of MK5 in PKA-triggered F-actin
remodelling.
Next, we have provided experimental evidence
that, similar to ERK3 and ERK4, co-expression of Calpha with MK5
stimulated the kinase activity of MK5 in vivo. We also found
enhanced phosphorylation of MK5 by in vitro kinase assay. Whether
this is due to increased phosphorylation of Thr-182 in the activation
loop of MK5 remains unsolved. We have tried to inspect whether
forskolin or Calpha trigger phosphorylation of MK5 at Thr-182 by
using two different commercially available antibodies
(anti-phospho-PRAK (Thr182) antibodies from Upstate, Charlottesville,
VA, USA, and anti-phospho-Threonine-Proline antibodies from Cell
signalling, Beverly, MA, USA). Unfortunately, none of these
antibodies worked in our hands, as they did not recognise in vitro
phosphorylated MK5 or immunoprecipitated MK5 from forskolin-treated
or Calpha -transfected cells. Moreover, we were unable to detect
PKA-induced phosphorylation of MK5 in vitro and in cell extracts with
a phospho-Ser/Thr PKA substrate antibody (Cell signalling
#9624).
The Calpha -MK5 interaction described here represents
a new example of crosstalk between the cAMP/PKA and MAPK signalling
pathways. Various branches of interaction between the cAMP/PKA
pathway and the different MAPK cascades have been demonstrated, e.g.
PKA-mediated regulation of Raf and p38 MAPK, and recently also of
another MAPKAPK, RSK1 [32,34,36,47].
Unphosphorylated RSK1 interacted with the regulatory RI subunits,
while activated RSK1 bound the catalytic subunits of PKA. The
interaction with PKA also regulated the subcellular localisation of
RSK1. The authors demonstrated that disruption of the interaction of
RI subunits with A-kinase PKA anchoring protein (AKAP) resulted in an
increase in cytosolic active RSK1 levels. The mechanism by which
PKA-induces nuclear export of MK5 appears to be different from RSK1
because we did not detect an in vivo interaction between MK5 and RI.
Chaturvedi and co-workers did not investigate whether Calpha
phosphorylated RSK1, nor was the effect of this interaction on the
intrinsic kinase activity of RSK1 tested [36].
We found that Calpha increased the phosphorylation levels of MK5, but
not vice versa.
Finally, we solved the biological relevance
for PKA-induced subcellular redistribution and activation of MK5 by
demonstrating that MK5 acted as a mediator for PKA-induced F-actin
remodelling. Cytoplasm-directed expression of a constitutive active
MK5 variant mimicked forskolin-induced F-actin rearrangement, while
ablation of MK5 expression by siRNA prevented forskolin-provoked
F-actin rearrangements. MK5 is not the only protein that is involved
in cytoskeletal architecture and whose subcellular location is
regulated by PKA. The RNA-binding protein,
polypyrimidine
tract-binding protein (PTB), was exported from the nucleus during
PKA-induced F-actin rearrangement/neurite outgrowth in PC12 cells.
Nuclear export of PTB required PKA-mediated phosphorylation of
Ser-16. PTB preferentially associated with beta -actin mRNA and
thwarting PTB expression by RNA interference disrupted PKA-, but not
NGF-induced neurite outgrowth in PC12 cells. Similar to MK5, PTB
represents a protein involved in cytoskeletal dynamics whose
subcellular distribution is regulated by PKA [48].
Several experimental observations indicate that p38 MAPK, MK2
and Hsp27 play a role in mediating alternations in filamentous actin
in response to stimuli. First, MK2, p38 MAPK, Hsp27 and Akt can form
a multiprotein complex [49,50]. In
addition,
stress-inducing stimuli activated p38 MAPK, and this coincided with
phosphorylation of MK2 and Hsp27, and changes in F-actin dynamics in
different cell lines [51-55] .
Furthermore, a regulatory role for
MK2 in F-actin dynamics during embryonic development was also
proposed by the work of Paliga and colleagues [56].
However, none of the above mentioned studies directly addressed the
role of MK5 in F-actin remodelling, nor was the participation of MK2
unequivocally proven by e.g. RNA interference studies for
overexpression of dominant negative variants of these kinases.
Studies in rat pulmonary artery microvascular endothelial cells
revealed a more direct causal involvement of MK2 in hypoxia-induced
actin reorganisation. Indeed, overexpression of a constitutive active
MK2 resembled, while a dominant negative MK2 mutant prevented the
effect of hypoxia on F-actin structure [57].
Stimuli-induced alterations in microfilament structure may require
different pathways that signal through distinct MKs. For example,
stimuli that activate p38 MAPK (e.g. oxidative stress) may engage
MK2, while stimuli signalling through the cAMP/PKA (e.g. secretin)
pathway may utilise MK5 to
provoke changes in F-actin structures
[51,55,58]. The
precise molecular mechanism by which MK5 participates in F-actin
remodelling remains elusive, but may in concordance with MK2 imply
phosphorylation of Hsp27. We could detect MK5-Hsp27 interaction by
co-immunoprecipitation and found that MK5 phosphorylated Hsp27 in
vitro (our unpublished results). On the other hand, studies by the
group of Gaestel with MK5 deficient mice jeopardise the role for MK5
in Hsp27 phosphorylation. The authors showed that Hsp27 did not
interact with MK5 in vivo, at least in primary murine cells isolated
from a strain with a mixed 129xC57/B6 genetic background
[22]. It cannot be ruled out that Hsp27 is
a bona fide
substrate for MK5 in rat PC12 cells or that PKA-provoked
phosphorylation/activation or conformational changes of MK5 are
required to enable in vivo interaction with Hsp27. Observations in
mk5-/- mice further support a putative role for MK5 in
regulation of the cytoskeleton structure and cell migration. Cell
motility requires reorganisation of the cytoskeleton and directed
cell migration is important for embryonic development. Depending on
the genetic background, MK5 deficient animals, but not mk2-/-
mice, displayed embryonic lethality with incomplete penetrance around
day E11.5. This developmental failure may be the result of impaired
cell migration [23].
Activation of the nuclear
transcription factor CREB by phosphorylation at Ser-133 has been
documented to be involved in cAMP-provoked cytoskeletal changes in
PC12 cells [59]. CREB can be
phosphorylated by
numerous protein kinases, including MAPKAPK [60]
.
It is unlikely that MK5-mediated F-actin rearrangements in
forskolin-treated PC12 cells requires the action of CREB because
forskolin induced redistribution of MK5 to another subcellular
compartment than CREB, and MK5 did not interact with CREB in vivo,
nor did MK5 phosphorylate CREB or activate CREB-mediated
transcription [61].
In conclusion, our
studies have revealed a novel function of MK5 in microfilament
rearrangements in response to increased cAMP levels and disclosed a
new interaction between the cAMP/PKA and the MAPK signalling
pathways. These findings may increase our molecular understanding of
stimulus-induced changes in microfilament and crosstalk between
signal transduction pathways. Whether MK5 is involved in other
cytoskeletal rearrangements and/or cell motility events awaits to be
proven, but phosphorylation of the putative MK5 substrate, Hsp25/27
also contributes to cell migration. In addition, the F-actin
interacting myosin light chain protein was shown to be an in vitro
substrate for MK5 [10,62-64].
Where, when, and under which
circumstances MK5 participates in cytoskeletal dynamics in an
organism are questions that need to be answered to increase our
understanding of MK5its role in normal and possible pathological
processes in order to apply therapeutic strategies in diseases
associated with perturbed MK5 activity.
Acknowledgements
We thank following colleagues for kindly providing
us with important reagents in this study: Dr. M. Gaestel (pEGFP-MK2),
Dr. G. Thiel (Cα-NLS
expression plasmid), Dr. B. Skålhegg (RIα
subunit antibodies), Dr. J. Saraste (PC12 cells), Dr R. A. Maurer
(PKI expression plasmid), Dr. S.H. Green (GPKAnes and GPKAnls
plasmids), Dr. J. Yao (PKA deficient PC12 cells), Division of Signal
Transduction Therapy, University of Dundee (ERK3 protein and
antibodies). The help of Linda Helander with the PC12 cells is
greatly appreciated. This work was supported by grants from the
Norwegian Research Council (NFR, project S5228), The Norwegian Cancer
Society (Kreftforeningen, project A01037), the Erna and Olav Aakres
Foundation, and the Family Blix Foundation.
Figures
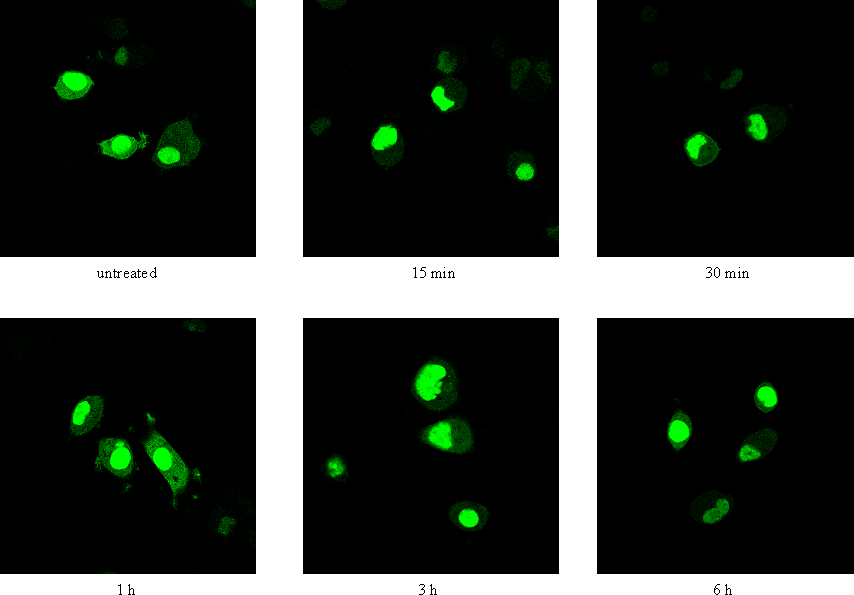
Figure 1 : Activation of the cAMP/PKA signalling pathway induces transient nuclear exclusion of MK5
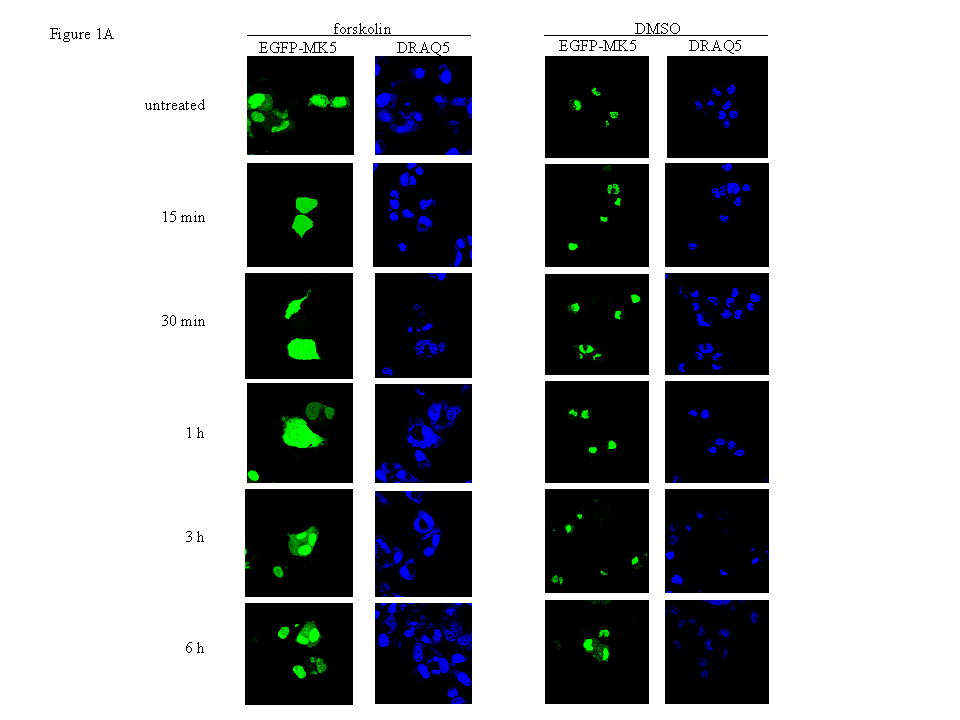
Figure 1A : PC12 cells were transfected with an expression plasmid for EGFP-MK5, and then stimulated with either 10 μM forskolin (FSK) or vehicle (DMSO) for the time points indicated. The subcellular localisation of the EGFP-MK5 fusion protein over time was visualised by confocal microscopy. Cell nuclei were visualised by DRAQ5 staining. The subcellular distribution of EGFP-MK5 was monitored by confocal microscopy. Fifty cells were counted and more than 80% displayed both nuclear and cytoplasmic presence of EGFP-MK5 after forskolin exposure.
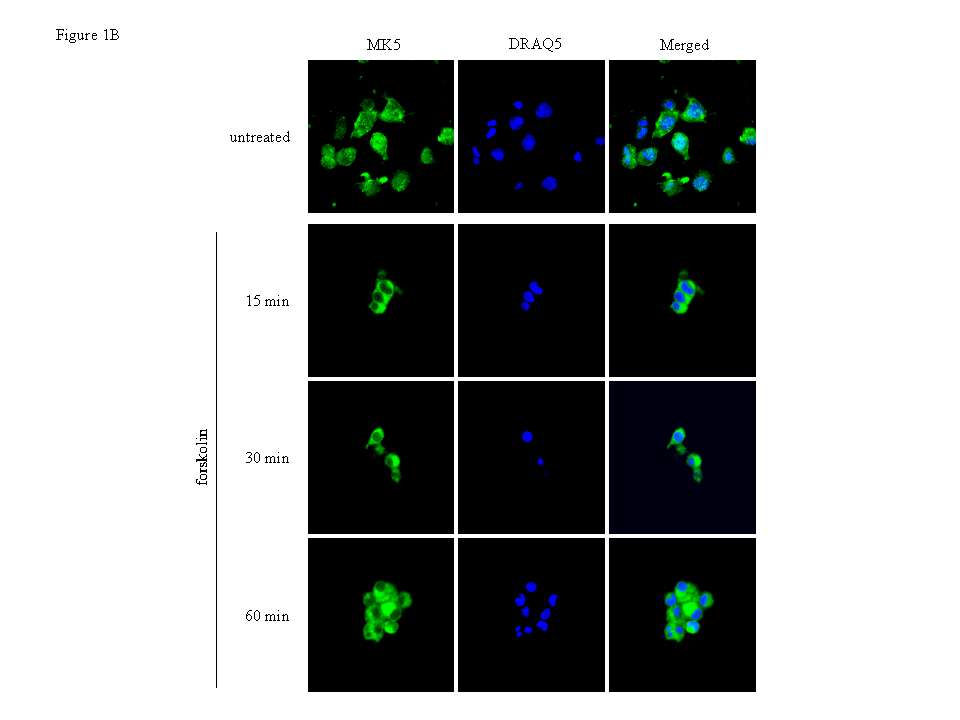
Figure 1B : PC12 cells were treated with 10 μM forskolin for the indicated time points and endogenous MK5 was visualised with anti-PRAK antibody (Santa Cruz) as primary antibody and Alexa 488 as secondary antibody. Nuclei were stained with DRAQ5.
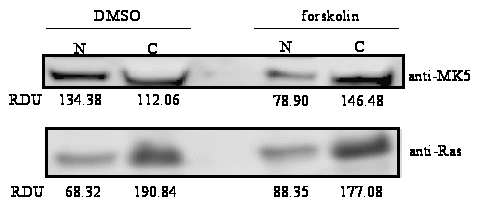
Figure 1C – First panel : PC12 cells were transfected with EGFP-MK5. Nuclear and cytoplasmic extracts were prepared from forskolin-treated cells (30 min) or vehicle- (DSMO) treated cells. The presence of EGFP-MK5 was assayed by western blotting. To ensure equal loading and to test for contamination between cytoplasmic and nuclear fractions, the membrane was re-probed with anti-Ras antibodies.
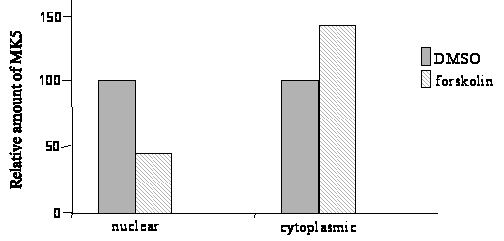
Figure 1C – Second panel : The intensity of the signals was determined by densitometry and the values are shown as relative densitometric units (RDU). Bottom panel: the ratio of RDUMK5:RDURAS in nuclear (respectively cytoplasmic) fraction of DMSO-treated cells was arbitrary set as 100% and the ratios of RDUMK5:RDURAS in the fractions of forskolin-treated cells was related to this.
Figure 2 : MK5, but not MK2, interacts with Cα in vivo
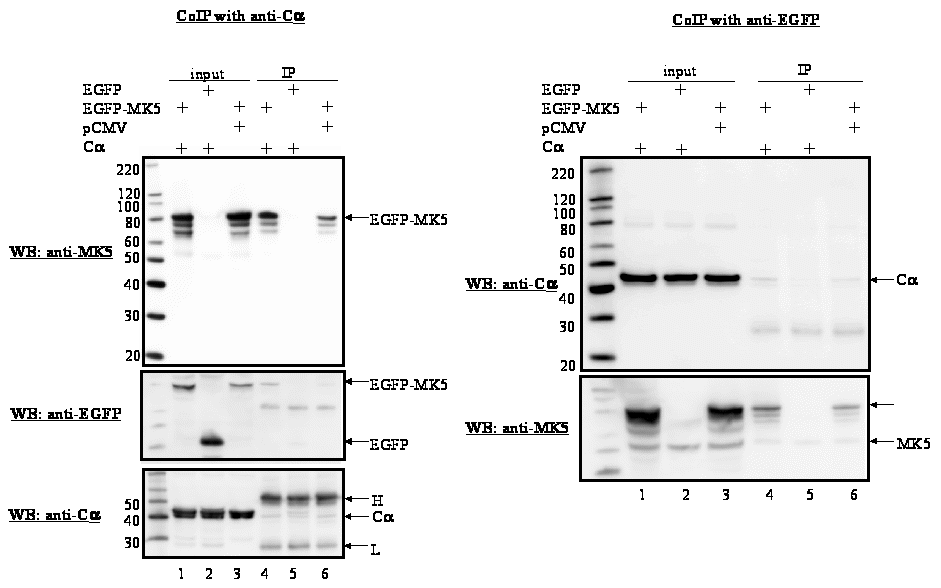
Figure 2A
:
HeLa cells were co-transfected with expression plasmids for Cα
and EGFP-MK5 or corresponding empty vectors (pCMV and pEGFP,
respectively). After 24 h, cells were lysed and Cα
was immunoprecipitated with anti-Cα
antibody (left panel) or EGFP-MK5 was immunoprecipitated with
anti-EGFP antibodies (right panel). The immunoprecipitated proteins
were separated by SDS-PAGE and western blotting was performed using
either anti-MK5 antibody (left panel) or anti-Cα antibody
(right panel). The coimmunoprecipitates with anti-Cα
were also assayed with anti-EGFP antibodies (middle part of left
panel). Lanes 1-3: input
control of the transfected cells; lanes
4-6:
immunoprecipitates. Lanes 1 and 4: cells transfected with expression
vectors for EGFP-MK5 and Cα;
lanes 2 and 5: cells
transfected with pEGFP vector and Cα
expression vector; lanes 3 and 6:
cells transfected with EGFP-MK5 and
empty vector for Cα
(pCMV). The positions of Cα,
MK5, the EGFP-MK5 fusion proteins, and the heavy (H) and light (L)
IgG chains are indicated. To
ascertain equal protein loading, the membranes were re-probed with an
antibody against the immunoprecipitated protein (bottom panel).
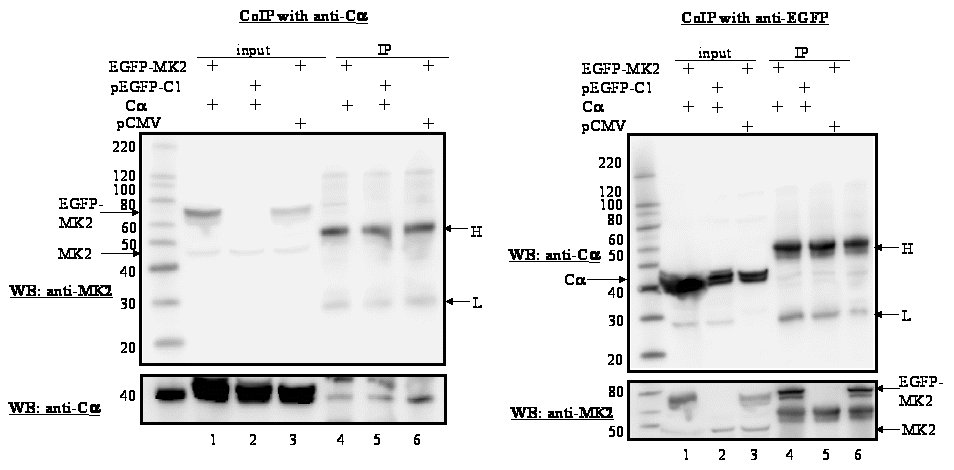
Figure 2.B: HeLa cells were co-transfected with expression plasmids for EGFP-MK2 and Cα or with the corresponding empty vectors (EGFP and pCMV, respectively). After 24 h, cells were lysed and Cα was immunoprecipitated with anti-Cα antibody (left panel) or EGFP-MK2 was immunoprecipitated with anti-EGFP antibodies (right panel). Lanes 1-3: input control of the transfected cells; lanes 4-6: western blotting of the immunoprecipitates with anti-Cα antibody. Lanes 1 and 4: cells transfected with expression vectors for EGFP-MK2 and Cα; lanes 2 and 5: cells transfected with empty vector for MK2 (EGFP) and Cα expression vector; lanes 3 and 6: cells transfected with MK2 expression plasmid and empty vector for Cα (pCMV). The molecular mass marker and the positions of MK2, MK2-EGFP fusion protein, and the heavy (H) and light (L) IgG chains are indicated. To ascertain equal protein loading, the membranes were re-probed with an antibody against the immunoprecipitated protein (bottom panel in the figure).
Figure 3 : The catalytic subunit Cα of PKA increased the phosphorylation level of MK5, but not MK2 in vitro

Figure
3A :
Top panel (IVK) shows in
vitro
phosphorylation of MK5 by Cα. Left lane:
purified Cα;
middle lane: purified MK5; right lane: Cα
plus MK5. The positions of phosphorylated Cα
(pCα)
and phosphorylated MK5 (pMK5) are indicated. Middle panel: the
samples used for in
vitro
kinase assay were tested for equal amounts of MK5 protein by western
blot with anti-MK5 antibodies. The values under the top and middle
panel represent relative densitometric units (RDU) of the scanned
phosphoMK5 (pMK5) and MK5 signals, respectively. The ratio of RDU of
total MK5 protein (western blot signals) and of phosphorylated MK5
(in
vitro
kinase signals) is shown in the bottom part.
Figure 3B Cα does not phosphorylate MK2 in vitro. Lane 1: MK2 protein; lane 2: purified Cα protein; lane 3: MK2 protein plus Cα; lane 4: myelin basic protein (MBP) plus Cα. The lower panels represent western blot with anti-MK2 and anti-Cα antibodies, respectively. Bovine serum albumin (BSA), which is present in the kinase assay buffer, is also phosphorylated by Cα. The positions of the phosphorylated proteins (pBSA, pCα, and pMBP) are shown. The amount of MK2 protein (middle panel) and Cα (bottom panel) in each sample was assayed by western blot.
Figure 4 : The PKA catalytic subunit Cα stimulates the kinase activity of MK5 in vitro and in vivo
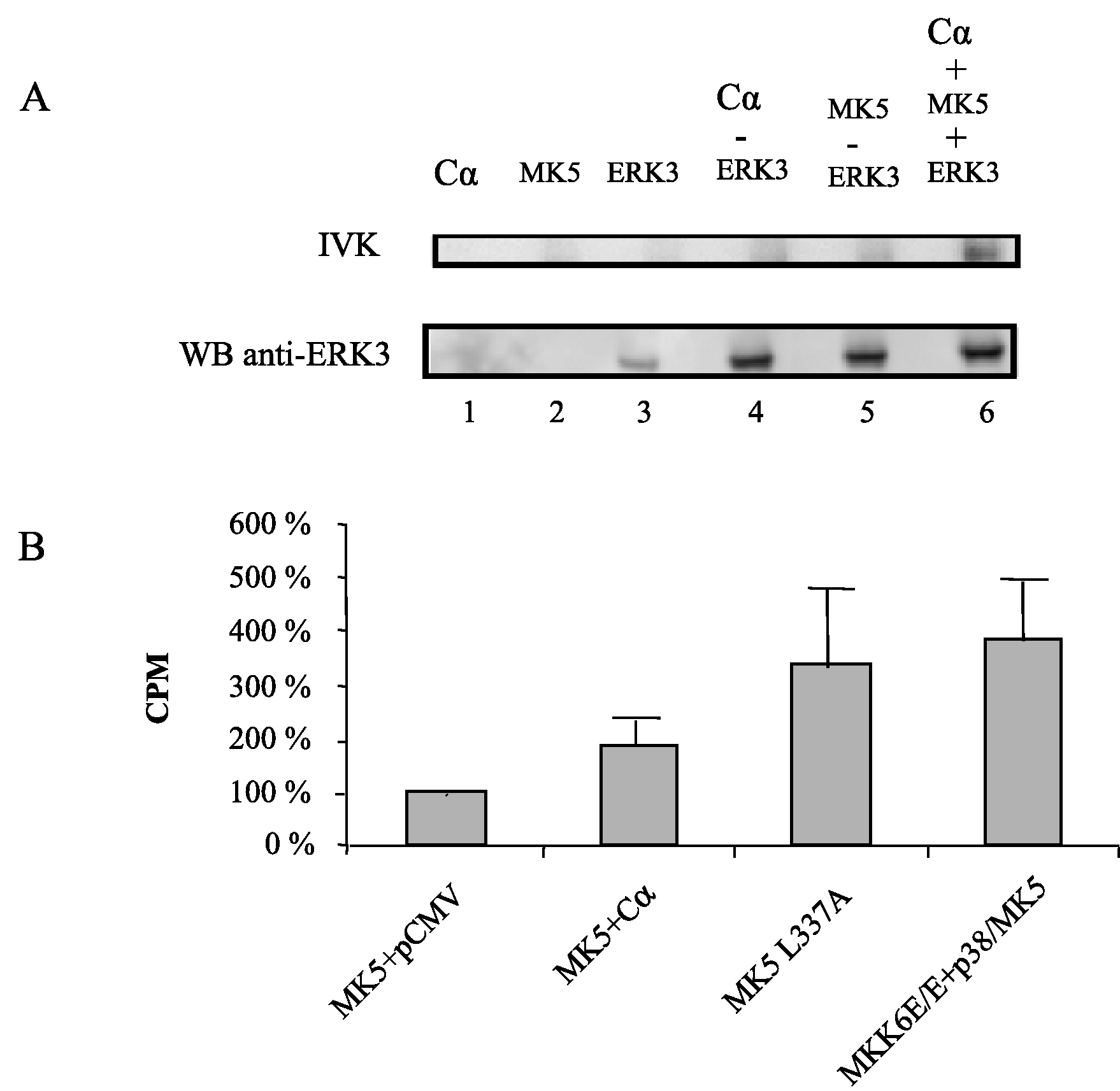
Figure 4B : Cells were transfected with expression plasmids for the EGFP-MK5 fusion protein and Cα or empty vector. As a positive control for the assay, cells were either transfected with plasmids encoding the constitutive active MK5 L337A mutant (EGFP-MK5 L337A) or with EGFP-MK5 and constitutive active MKK6E/E plus p38 MAPK. Cell lysates were immunoprecipitated with anti-EGFP antibody and the MK5 kinase activity was determined by an immune complex kinase assay with PRAKtide as a substrate. The relative kinase activity in of wild-type MK5 in the absence of Cα was arbitrary set as 100% and the activity of the other samples was related to this. The results represent the average of 5 to 6 independent experiments.
Figure 5 : Forskolin- or Cα-induced nuclear export of MK5 requires nuclear entrance of Cα
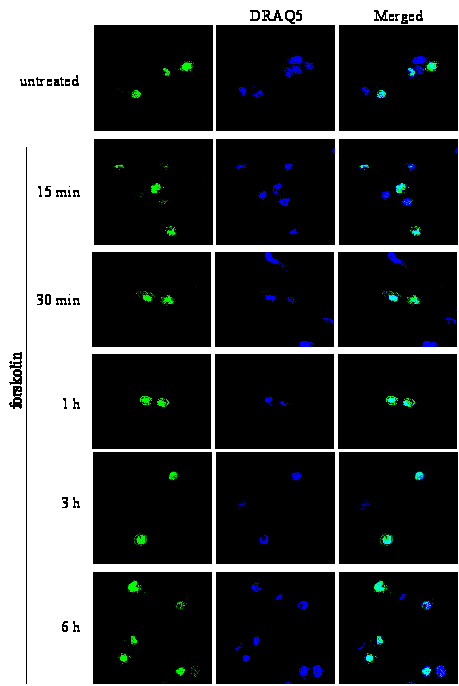
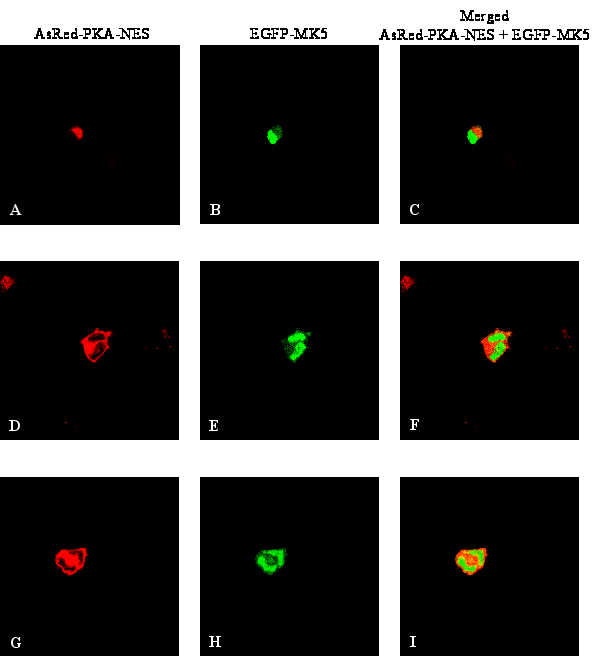
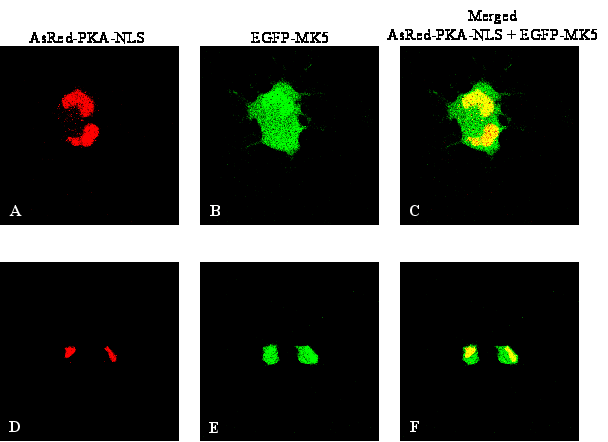
Figure 6 : The kinase activities of both the PKA catalytic subunit Cα and MK5 are required to induce nuclear export of MK5
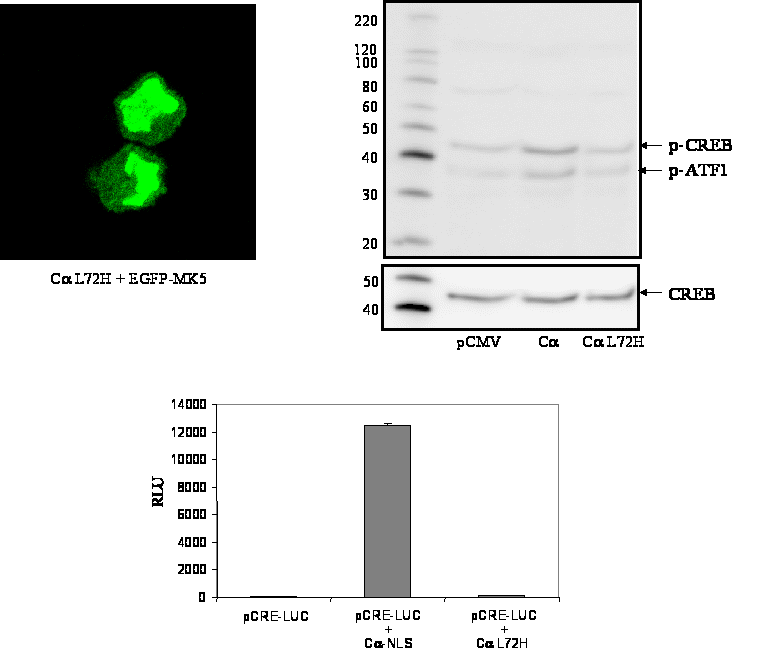
Figure 6A : PC12 cells were co-transfected with expression plasmids encoding EGFP-MK5 fusion protein and with nucleus-directed kinase dead Cα L72H mutant. The subcellular distribution of MK5 was monitored by confocal microscopy (left panel on the top). Middle panel on the top: phosphospecific CREB antibodies failed to detect increased phosphorylation of CREB at Ser-133 in cells expressing kinase dead Cα L72H, while phosphoCREB was monitored in cells expressing kinase active Cα. Lane 1: extracts of cells transfected with empty vector pRcCMV; lane 2: extracts of cells transfected with wild-type Cα expression vector; lane 3: extracts of cells transfected with expression plasmid for Cα L72H. The cells were serum-starved overnight after transfection before they were harvested for western blotting analysis. The phosphoCREB antibodies cross-react with phospho-ATF1, a member of the CREB superfamily. The membrane was stripped and blotted with anti-CREB antibodies to ensure equal loading. The molecular mass (in kDa) of the protein marker is indicated. Right panel on the top: HEK293 cells were transfected using Lipofectamine 2000 with 100 ng pCRE-LUC reporter plasmid and 100 ng of expression vector encoding wild-type Cα or kinase dead Cα L72H. Four hours after transfection, the medium was replaced with fresh medium containing 0.2% serum. Luciferase activity (expressed as relative luciferase units; RLU) was measured 20 hrs later. The results represent the average +/- SD of three independent experiments.
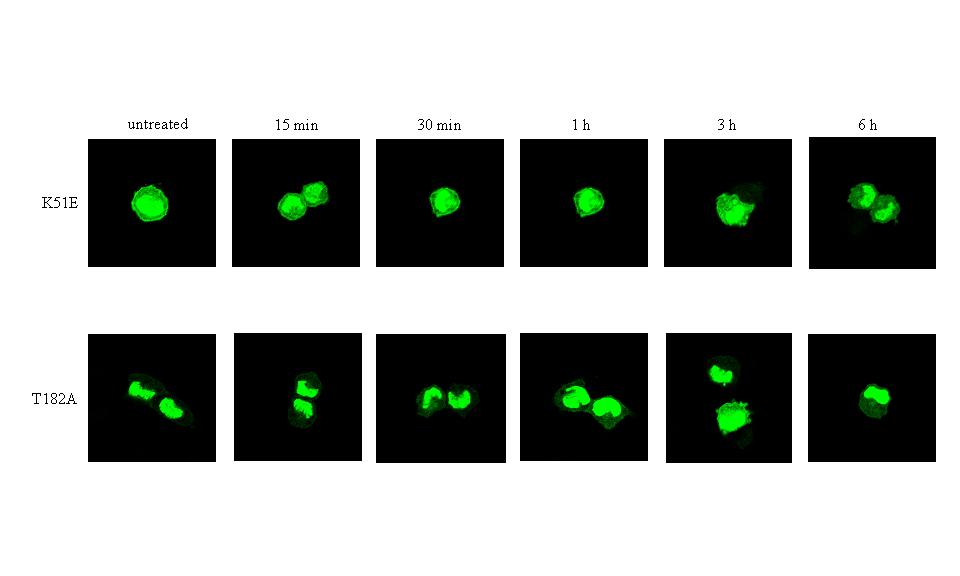
Figure 6B : Forskolin does not induce transient nuclear exclusion of the kinase death MK5 K51E and MK5 T182A mutants. Cells were transfected with expression plasmids for the EGFP-MK5 K51E (top panel) or EGFP-MK5 T182 (bottom panel) fusion proteins. Green fluorescence was monitored at different time points after stimulation with 10 μM forskolin.
Figure 7 : Forskolin alters the amount of F-actin and PKA-induced F-actin remodeling is mediated by MK5
Figure
7B :
Nuclear exclusion of activated PKA by PKI prevents forskolin-induced
F-actin remodeling. Cells transfected with a PKI expression plasmid
were stimulated with forskolin 24 hrs after transfection and were
then monitored for microfilament structure by staining F-actin.
Figure 7C : PC12 cells, transfected with scrambled siRNA, were treated with forskolin (10 μM) for the indicated time points and the architecture of the microfilaments was examined by staining F-actin with Alexa Fluor 594 phalloidin.
Figure 7D : Depletion of MK5 protein ablates forskolin-induced F-actin rearrangements in PC12 cells. Cells were transfected with 100 μM siRNA/106 cells and were stained for F-actin 48 hrs after transfection.
Supplementary Data
Figure 1

Figure 2
Bibliography
1. Huang, C., Jacobson, K., and Schaller, M.D. (2004) J. Cell Sci. 117, 4619-4628
2. Roux, P.P., and Blenis, J. (2004) Microbiol. Mol. Biol. Rev. 68, 320-344
3. Imajo, M., Tsuchiya, Y., and Nishida, E. (2006). IUBMB Life 58, 312-317
4. Krens, S.F., Spaink, H.P., and Snaar-Jagalska B.E. (2006) FEBS Lett. 580, 4984-4990
5. Whitmarsh, A.J. (2007) Biochim Biophys Acta. 1773, 1285-1298
6. Manning, G., Whyte, D.B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002) Science 298, 1912-1934
7. Stokoe, D., Campbell, D.G., Nakielny, S., Hidaka, H., Leevers, S.J., Marshall, C., and Cohen, P. (1992) EMBO J. 11, 3985-3994
8. Sithanandam, G., Latif, F., Duh, F.M., Bernal, R., Smola, U., Li, H., Kuzmin, I., Wixler, V., Geil, L. and Shrestha, S. (1996) Mol. Cell. Biol. 16, 868-876
9. New, L., Jiang, Y., Zhao, M., Liu, K., Zhu, W., Flood, L.J., Kato, Y., Parry, G.C.N., and Han, J. (1998) EMBO J. 17, 3372-3384
10. Ni, H., Wang, X.S., Diener, K., and Yao, Z. (1998) Biochem. Biophys. Res. Commun. 243, 492-496
11. Gaestel, M. (2006) Nature Rev. Mol. Cell Biol. 7, 120-130
12. Gerits, N., Kostenko, S., and Moens, U. (2007) Transgenic Res. 16, 281-314
13. Han, J., and Ulevitch, R.J. (1999) Nature Cell Biol. 1, E39-40
14. Kotlyarov, A., Neininger, A., Schubert, C., Eckert, R., Birchmeier, C., Volk, H.D., and Gaestel, M. (1999) Nature Cell Biol. 1, 94-97
15. Kotlyarov, A., Yannoni, Y., Fritz, S., Laas, K., Telliez, J.B., Pitman, D., Lin, L.L., and Gaestel, M. (2002) Mol. Cell. Biol. 22, 4827-4835
16. Manke I.A., Nguyen, A., Lim, D., Stewart, M.Q., Elia, A.E.H., and Yaffe, M.B. (2005) Mol. Cell, 17, 37-48
17. Culbert, A.A., Skaper, S.D., Howlett, D.R., Evans, N.A., Facci, L., Soden, P.E., Seymour, Z.M., Guillot, F., Gaestel, M. , and Richardson, J.C. (2006) J. Biol. Chem. 281, 23658-23667
18. Kobayashi, M., Nishita, M., Mishima, T., Ohashi, K., and Mizuno, K. (2006) EMBO J. 25,713-726
19. Neufeld, B., Grosse-Wilde, A., Hoffmeyer, A., Jordan B.W., Chen, P., Dinev, D., Ludwig, S. and Rapp, U.R. (2000) J. Biol. Chem. 275, 20239-20242
20. Voncken, J.W., Niessen, H., Neufeld, B., Rennefahrt, U., Dahlmans, V., Kubben, N., Holzer, B., Ludwig, S., and Rapp, U.R. (2005) J. Biol. Chem. 280, 5178-5187
21. Houben, R., Becker, J.C., and Rapp, U.R. (2005) J. Carcinogen. 20, 23
22. Shi, Y., Kotlyarov, A., Laas, K., Gruber, A.D., Butt, E., Marcus, K., Meyer, H.E., Friedrich, A., Volk, H.D., and Gaestel, M. (2003) Mol. Cell. Biol. 23, 7732-7741
23. Schumacher, S., Laass, K., Kant, S., Shi, Y., Visel, A., Gruber, A.D., Kotlyarov, A., and Gaestel, M. (2004) EMBO J. 23, 4770-4779
24. Seternes, O.M., Mikalsen, T., Johansen, B., Michaelsen, E., Armstrong, C.G., Morrice, N.A., Turgeon, B., Meloche, S., Moens, U., and Keyse, S.M. (2004) EMBO J. 23, 4780-4791
25. Kant, S., Schumacher, S., Singh, M.K., Kisper, A., Kotlyarov, A., and Gaestel, M. (2006) J. Biol. Chem. 281, 35511-35519
26. Åberg, E., Perander, M., Johansen, B., Julien, C., Meloche, S., Keyse, S.M., and Seternes, O.M. (2006) J. Biol. Chem. 281, 35499-35510
27. Sun, P., Yoshizuka, N., New, L., Moser, B.A., Li, Y., Liao, R., Xie, C., Chen, J., Deng, Q., Yamout, M., Dong, M.Q., Frangou, C.G., Yates III, J.R., Wright, P.E., and Han, J. (2007) Cell 128, 295-308
28. Skålhegg, B., and Taskén, K. (2000) Front. Biosci. 5, D678-693
29. Stork, P.J., and Schmitt, J.M. (2002) Trends Cell Biol. 12, 258-266
30. Howe, A.K. (2004) Biochim. Biophys. Acta 1692, 159-174
31. Johannessen, M., Moens, U. (2005) Trends in Cellular Signalling, 41-78. Nova Science Publishers, NY
32. Saxena, M., Williams, S., Taskén, K., and Mustelin, T. (1999) Nature Cell Biol. 1, 305-311
33. Delghandi, M.P., Johannessen, M., and Moens, U. (2005) Cell Signal. 17, 1343-1351
34. Houslay, M.D. (2006) Sci STKE. 349, pe32
35. Pearson, G.W., Earnest, S., and Cobb, M.H. (2006) Mol. Cell. Biol. 26, 3039-3047
36. Chaturvedi, D., Poppleton, H.M., Stringfield, T., Barbier, A., and Patel, T.B. (2006) Mol Cell. Biol. 26, 4586-4600
37. Seternes, O.M., Johansen, B., Hegge, B., Johannessen, M., Keyse, S.M. and Moens, U. (2002) Mol. Cell. Biol. 22, 6931-6945
38. Day, R.N., Walder, J.A., and Maurer, R.A. (1989) J. Biol. Chem. 264, 431-436
39. Thiel, G., Al Sarraj, V., Vinson, C., Stefano, L., and Bach, K. (2005) J. Neurochem. 92, 321-336
40. Thyssen, G., Li, T.H., Lehmann, L., Zhuo, M., Sharma, M., and Sun, Z. (2006) Mol. Cell. Biol. 26, 8857-8867
41. Bok, J., Zha, X.M., Cho, Y.S., and Green, S.H. (2003) J. Neurosci. 23, 777-787
42. Johannessen, M., Delghandi, M.P., Seternes, O.M., Johansen, B. and Moens, U. (2004) Cell Signal. 16, 1187-1199
43. New, L., Jiang, Y., and Han, J. (2003) Mol. Biol. Cell 14, 2603-2616
44. Yao, J., Erickson, J.D., and Hersch, L.B. (2004) Traffic 5, 1006-1016
45. Dalton, G.D. and Dewey, W.L. (2006) Neuropeptides 40, 23-34
46. Montminy, M. (1997) Annu. Rev. Biochem. 66, 807-822
47. Dumaz, N., and Marais R. (2005) FEBS J. 272, 3491-504
48. Ma, S., Liu, G., Sun, Y., Xie, J. (2007) Biochim Biophys. Acta 1773, 912-923
49. Zheng, C., Lin, Z., Zhao, Z.J., Yang, Y., Niu, H. and Shen, X. (2006) J. Biol. Chem. 281, 37215-37226
50. Wu, R., Kausar, H., Johnson, P., Montoya-Durango, D.E., Merchant, M. and Rane, M.J. (2007) J. Biol. Chem. 282, 21598-21608
51. Huot J., Houle, F., Marceau, F., and Landry, J. (1997) Circ. Res. 80, 383-392
52. Larsen, J.K., Yamboliev, I.A., Weber, L.A., and Gerthoffer, W.T. (1997) Am. J. Physiol. 273, L930-L940
53. Huot, J., Houle, F., Rousseau, S., Deschesnes, R.G., Shah, G.M., and Landry, J. (1998) J. Cell Biol. 143, 1361-1373
54. Schäfer, C., Ross, S.E., Bragado, M.J., Groblewski, G.E., Ernst, S.A., and Williams, J.A. (1998) J. Biol. Chem. 273, 24173-24180
55. Azuma, N., Akasaka, N., Kito, H., Ikeda, M., Gahtan, V., Sasajima, T. and Sumpio, B.E. (2001) Am. J. Physiol. Heart Circ. Physiol. 280, H189-197
56. Paliga, A.J.M., Natale, D.R., and Watson, A.J. (2005) Biol. Cell 97, 629-640
57. Kayyali, U.S., Pennella, C.M., Trujillo, C., Villa, O., Gaestel, M., and Hassoun, P.M. (2002) J. Biol. Chem. 277, 42596-42602
58. Kim, H.S., Yumkham, S., Kim, S.H., Yea, K., Shin, Y.C., Ryu, S.H., and Suh, P.G. (2006) Exp. Mol. Med. 38, 85-93
59. Shimomura, A., Okamoto, Y., Hirata, Y., Kobayashi, M., Kawakami, K., Kiuchi, K., Wakabayashi, T., and Hagiwara, M. (1998) J. Neurochem. 70, 1029-1034
60. Johannessen, M., and Moens, U. (2007) Front Biosci. 12, 1814-1832
61. Johannessen, M., Delghandi, M.P., Rykx, A., Dragset, M., Vandenheede, J.R., Van Lint, J., and Moens, U. (2007) J. Biol. Chem. 282, 14777-14787
62. Hedges, J.C., Dechert, M.A:, Yamboliev, I.A., Martin, J.L., Hickey, E., Weber, L.A., and Gerthoffer, W.T. (1999) J. Biol. Chem. 274, 24211-24219
63. Pichon, S., Bryckaert, M., and Berrou, E. (2004) J. Cell Sci. 117, 2569-2577
64.
Rousseau, S., Dolado, I., Beardmore, V., Shpiro, N., Marquez, R.,
Nebrada, A.R., Arthur, J.S., Case, L.M., Tessier-Lavigne, M.,
Gaestel, M., Cuenda, A., and Cohen, P. (2006) Cell
Signal.
18,
1897-1905
Footnotes
- 1
- Abbreviations: Cα, catalytic subunit PKA isoform α; CRE, cAMP-response element; DMSO, dimethyl sulfoxide; EGFP, enhanced green fluorescence protein; MAPK, mitogen-activated protein kinase; MAPKAPK, MAPK-activated protein kinase; MBP, myelin basic protein; MK, MAPKAPK; NES, nuclear export signal; NLS, nuclear localisation signal; PKA, cAMP-dependent protein kinase; PTB, polypyrimidine tract-binding protein; RI, regulatory subunit PKA type I.
About this document ...
Modulation of F-actin rearrangements by the cyclic AMP/PKA pathway is mediated by MAPKAP Kinase 5 and requires PKA-induced nuclear export of MK5
This document was generated using the LaTeX2HTML translator Version 2002-2-1 (1.71)
Copyright © 1993, 1994, 1995, 1996, Nikos Drakos, Computer
Based Learning Unit, University of Leeds.
Copyright © 1997,
1998, 1999, Ross Moore,
Mathematics Department, Macquarie University, Sydney.
The translation was initiated on 2007-12-21