De L-929 cellijn is een fibroblastcellijn die men in Rouxflessen laat
groeien totdat men een monolaag bekomt. Het is een continue cellijn
die afkomstig is van normaal subcutaan areolair en adipeus weefsel
van een 100 dagen oude mannelijke (C3H/An) muis [54].
De cellen worden gecultiveerd in MEM (Invitrogen Corporation) (zie
bijlage A.1) aangerijkt met 10% paardenserum in
de incubator op 37^C bij een 5% ![]() -druk. De cellen werden oorspronkelijk
aangekocht bij het American Type Culture Collection (ATCC).
-druk. De cellen werden oorspronkelijk
aangekocht bij het American Type Culture Collection (ATCC).
Omdat zich op een bepaald moment een probleem van bacteriële contaminatie
voordeed, werd er beslist om 2 nieuwe antimicrobiële stoffen toe te
voegen aan het celmedium. De eindconcentratie van deze antimicrobiële
stoffen in het medium bedroeg 0.54 mM gentamycine (Invitrogen Corporation)
en 2,7 ![]() M Fungizone
M Fungizone![]() of amphotericine B (Invitrogen
Corporation).
of amphotericine B (Invitrogen
Corporation).
Gentamycine is een aminoglycoside dat vooral micromonospora en pseudomonas aeroginosa aantast. Dit gebeurt door een irreversiebele inhibitie van de proteïnensynthese. Resistentie kan verworven worden als er extrachromosomaal gecontroleerde enzymen aanwezig zijn (de R-factoren) die het antibiotica inactiveren door het te fosforyleren, te acetyleren of door er een adenylatie op uit te voeren [11].
Fungizone![]() of amphotericine B bindt op
sterolen (ergosterol) aanwezig in de celmembraan van sommige fungi,
algen, protozoa en dierlijke cellen. De binding zorgt voor een stijging
van de membraanpermeabiliteit, zodat essentiële componenten (bv. natrium)
naar het extracellulair milieu lekken [11].
of amphotericine B bindt op
sterolen (ergosterol) aanwezig in de celmembraan van sommige fungi,
algen, protozoa en dierlijke cellen. De binding zorgt voor een stijging
van de membraanpermeabiliteit, zodat essentiële componenten (bv. natrium)
naar het extracellulair milieu lekken [11].
Om te weten hoeveel virus we moeten toevoegen om de cellen te infecteren met 10 p.f.u. (zie sectie 2.2) per cel, moet het aantal cellen gekend zijn. Daarvoor nemen we een petrischaal met cellen en voegen aan deze schaal 2 ml trypsine toe. Deze (verdunnings)factor wordt in rekening gebracht, net als een factor die eigen is aan de Bürkerkamer (= 5000). We tellen het aantal cellen in de Bürkerkamer binnen 50 vierkantjes m.b.v. een telapparaatje. Dit getal zal dienen als referentie voor de rest van de populatie. We bekomen dan bijvoorbeeld: 227 cellen x 2 ml trypsine x 5000 = 2 270 000 cellen. Omdat de titer van het virus bekend is en de cellen geïnfecteerd worden aan bv. 10 p.f.u./cel, weten we hoeveel microliter virussuspensie we per petrischaal moeten toevoegen. We infecteren aan 10 p.f.u./cel omdat de cellen dan vanaf het begin allemaal geïnfecteerd raken.
Voor onze experimenten gebruiken we de DA-stam van het TMEV (zie sectie 1.2). Deze demyeliniserende stam van het TMEV werd bekomen bij Prof. Dr. T. Michiels (UCL). De L-cel is een uitstekende waardcel voor dit virus.
Voor een viruskweek gebruiken we ongeveer 20 Rouxflessen (150 c![]() )
met L-cellen waarvan het medium werd afgegoten. De cellen werden nagespoeld
met een weinig medium of trisbuffer (bijlage A.2).
Hierop kweken we de DA-stam van het TMEV door 2 ml P3- materiaal (P
= passages) per Rouxfles op de cellen te brengen en 1 uur te laten
adsorberen. We kantelen de Rouxflessen elke 15 minuten. Tenslotte
voegen we overal 15 ml medium, dat 1% paardenserum bevat, toe.
)
met L-cellen waarvan het medium werd afgegoten. De cellen werden nagespoeld
met een weinig medium of trisbuffer (bijlage A.2).
Hierop kweken we de DA-stam van het TMEV door 2 ml P3- materiaal (P
= passages) per Rouxfles op de cellen te brengen en 1 uur te laten
adsorberen. We kantelen de Rouxflessen elke 15 minuten. Tenslotte
voegen we overal 15 ml medium, dat 1% paardenserum bevat, toe.
Na het optreden van het cytopathogeen effect (= morfologische
veranderingen die de cel ondergaat bij virale infectie zoals marginatie
van het chromatine, membraanvesikels in het cytoplasma, membraanlekken,.. [29])
wordt het viraal materiaal geoogst door het te onderwerpen aan 3 vriesdooistappen.
Hierbij bevriest men het virus gedurende 2 uur bij -80^C om het daarna
te ontdooien bij 4^C. Deze procedure herhaalt men 2 keer en is nodig
om het resterende materiaal los te krijgen. Daarna brengen we de inhoud
van de Rouxflessen over naar centrifugatiebuizen en centrifugeren
deze gedurende 10 minuten bij 3000 t.p.m. (1000xG) (Super Minor centrifuge
[MSE]). Dit zorgt voor een scheiding van het viraal materiaal
(dat in het supernatans terechtkomt) en het cellulair materiaal (dat
gepelleteerd wordt). Het supernatans ondergaat daarna een ultracentrifugatiestap
in een TFT 70.38 rotor gedurende 1 uur 30 minuten aan 40 000 t.p.m.
(130 000xG) bij 4^C. Hierbij wordt het viraal materiaal gepelleteerd,
zodat men het supernatans kan verwijderen, waardoor men het virus
concentreert. Men resuspendeert het pellet in 250 ![]() l trisbuffer
en bepaalt hiervan de titer via plaquetitratie (zie sectie 2.2).
l trisbuffer
en bepaalt hiervan de titer via plaquetitratie (zie sectie 2.2).
Met deze techniek bepaalt men het aantal infectieuze partikels in een virale suspensie. Dit doet men door gevoelige cellen in een petrischaal tot een monolaag te laten groeien en ze daarna in contact te brengen met verschillende verdunningen van het virus. Ten gevolge van de infectie neemt men plaques (= zones van celdestructie te wijten aan in vitro virusinfectie) waar. De Plaque Forming Unit (p.f.u.) is dus een maat voor het aantal infectieuze virusdeeltjes waarbij elke plaque overeenkomt met de hoeveelheid cellyse, die oorspronkelijk geïnduceerd werd door 1 viraal partikel.
We maken een tienvoudige verdunningsreeks van het virus in trisbuffer
(pH 7.4) (zie bijlage A.2), vertrekkende van de
verdunning 10![]() tot 10
tot 10![]() via de meesleeptechniek. Deze
verdunningsreeks kan, afhankelijk van de vereisten, aangepast worden.
via de meesleeptechniek. Deze
verdunningsreeks kan, afhankelijk van de vereisten, aangepast worden.
De petrischaaltjes (diameter = 55 mm) met L-929 cellen worden nu
uit de incubator gehaald. Het medium wordt weggezogen en op elk schaaltje
wordt 100 ![]() l van een bepaalde verdunning gebracht. Er wordt
1 schaaltje ter controle gebruikt. Dit zal 100
l van een bepaalde verdunning gebracht. Er wordt
1 schaaltje ter controle gebruikt. Dit zal 100 ![]() l trisbuffer
bevatten. We verdelen de vloeistof volledig over het oppervlak van
de petrischaaltjes door ze te kantelen en plaatsen ze 15 minuten in
de incubator zodat het virus kan adsorberen. Daarna kantelen we de
plaatjes opnieuw. We herhalen deze procedure 3 keer. Gedurende de
laatste 15 minuten maken we het overlaymedium klaar. Hiervoor
gebruiken we steriele agar (1.6%) die we laten smelten. Daarna koelen
we deze agar (bekomen bij Difco) af omdat de cellen en het virus bij
het opbrengen van een te hete agarlaag zullen degraderen. Voor het
overlaymedium gebruiken we 50 delen agar (een colloïdale gelatineuze
stof die wordt toegevoegd om de mobiliteit van het virus te beperken
tot zijn naaste omgeving), 50 delen dubbelgeconcentreerd medium en
1% paardenserum. We gebruiken eenzelfde volume (20 ml) dubbelgeconcentreerd
medium (MEM2x, bijlage A.1) omdat de agar voor een
verdunningsfactor zorgt en de cellen nog 3 dagen in optimale conditie
moeten verkeren vooraleer ze gefixeerd worden. In het recipiënt komen
dus achtereenvolgens het medium, het serum en de agar. We vortexen.
De petrischaaltjes worden uit de incubator gehaald en er wordt telkens
5 ml overlaymedium op de schaaljes gebracht. We laten deze nu 3 dagen
incuberen in een
l trisbuffer
bevatten. We verdelen de vloeistof volledig over het oppervlak van
de petrischaaltjes door ze te kantelen en plaatsen ze 15 minuten in
de incubator zodat het virus kan adsorberen. Daarna kantelen we de
plaatjes opnieuw. We herhalen deze procedure 3 keer. Gedurende de
laatste 15 minuten maken we het overlaymedium klaar. Hiervoor
gebruiken we steriele agar (1.6%) die we laten smelten. Daarna koelen
we deze agar (bekomen bij Difco) af omdat de cellen en het virus bij
het opbrengen van een te hete agarlaag zullen degraderen. Voor het
overlaymedium gebruiken we 50 delen agar (een colloïdale gelatineuze
stof die wordt toegevoegd om de mobiliteit van het virus te beperken
tot zijn naaste omgeving), 50 delen dubbelgeconcentreerd medium en
1% paardenserum. We gebruiken eenzelfde volume (20 ml) dubbelgeconcentreerd
medium (MEM2x, bijlage A.1) omdat de agar voor een
verdunningsfactor zorgt en de cellen nog 3 dagen in optimale conditie
moeten verkeren vooraleer ze gefixeerd worden. In het recipiënt komen
dus achtereenvolgens het medium, het serum en de agar. We vortexen.
De petrischaaltjes worden uit de incubator gehaald en er wordt telkens
5 ml overlaymedium op de schaaljes gebracht. We laten deze nu 3 dagen
incuberen in een ![]() -oven (5%
-oven (5% ![]() op 37^C). Daarna nemen
we een 10%-ige formaldehyde-oplossing en brengen op elk schaaltje
5 ml formaldehyde. Dit is nodig om de monolayer te fixeren zodat deze
er niet afkomt bij het verwijderen van de agar. We laten het fixatief
35 à 40 minuten inwerken. Daarna zuigen we de formaldehyde eraf en
verwijderen de agar voorzichtig met een spatel zodat de monolayer
niet wordt aangetast. Tenslotte brengen we 3 ml kristalviolet-oplossing2.1 (10% verdunde oplossing van 10 g kristalviolet, 100 ml ethanol
en 400 ml
op 37^C). Daarna nemen
we een 10%-ige formaldehyde-oplossing en brengen op elk schaaltje
5 ml formaldehyde. Dit is nodig om de monolayer te fixeren zodat deze
er niet afkomt bij het verwijderen van de agar. We laten het fixatief
35 à 40 minuten inwerken. Daarna zuigen we de formaldehyde eraf en
verwijderen de agar voorzichtig met een spatel zodat de monolayer
niet wordt aangetast. Tenslotte brengen we 3 ml kristalviolet-oplossing2.1 (10% verdunde oplossing van 10 g kristalviolet, 100 ml ethanol
en 400 ml ![]() ) op de schaaltjes en laten deze gedurende ongeveer
30 minuten de levende cellen kleuren. Op die manier worden de plaques
zichtbaar als niet-gekleurde regio's. We spoelen de petrischalen met
) op de schaaltjes en laten deze gedurende ongeveer
30 minuten de levende cellen kleuren. Op die manier worden de plaques
zichtbaar als niet-gekleurde regio's. We spoelen de petrischalen met
![]() en laten ze aan de lucht drogen. Tenslotte kunnen de plaques
geteld worden en kan de virustiter bepaald worden, rekening houdend
met de verdunningsfactor en de hoeveelheid virussuspensie opgebracht.
en laten ze aan de lucht drogen. Tenslotte kunnen de plaques
geteld worden en kan de virustiter bepaald worden, rekening houdend
met de verdunningsfactor en de hoeveelheid virussuspensie opgebracht.
We voegen ![]() S L-methionine toe aan de geïnfecteerde cellen
met als doel het virus te merken. Dit aminozuur wordt immers in de
proteïnen geïncorporeerd tijdens de proteïnensynthese. Om een efficiënte
opname van het toegevoegde isotoop te verzekeren, worden de cellen
eerst uitgeput aan methionine.
S L-methionine toe aan de geïnfecteerde cellen
met als doel het virus te merken. Dit aminozuur wordt immers in de
proteïnen geïncorporeerd tijdens de proteïnensynthese. Om een efficiënte
opname van het toegevoegde isotoop te verzekeren, worden de cellen
eerst uitgeput aan methionine.
We brengen 100 ![]() l virussuspensie op de cellen en laten het 1
uur adsorberen bij 37^C in een incubator met 5%
l virussuspensie op de cellen en laten het 1
uur adsorberen bij 37^C in een incubator met 5% ![]() , waarbij
we de schaaltjes elke 15 minuten kantelen. De controle voorzien we
met 100
, waarbij
we de schaaltjes elke 15 minuten kantelen. De controle voorzien we
met 100 ![]() l trisbuffer. Ondertussen maken we het medium klaar.
Dit bestaat uit MEM (zie bijlage A.1) zonder
methionine, maar met 1mM natriumpyruvaat- en een 2mM glutaminesupplement.
Standaard brengen we 850
l trisbuffer. Ondertussen maken we het medium klaar.
Dit bestaat uit MEM (zie bijlage A.1) zonder
methionine, maar met 1mM natriumpyruvaat- en een 2mM glutaminesupplement.
Standaard brengen we 850 ![]() l medium op de cellen.
l medium op de cellen.
We plaatsen de petrischalen terug in de incubator totdat het cytopathogeen
effect optreedt (ongeveer 5 uur p.i.). Dan voegen we 50 ![]() l Tran
l Tran
![]() label (ICN Biomedicals) toe. Dit label is afkomstig van
een cellulair hydrolysaat van E. Coli dat meer dan 70% methionine
bevat. Het bevat ook andere gemerkte (bv. cystine) en niet-gemerkte
componenten.
label (ICN Biomedicals) toe. Dit label is afkomstig van
een cellulair hydrolysaat van E. Coli dat meer dan 70% methionine
bevat. Het bevat ook andere gemerkte (bv. cystine) en niet-gemerkte
componenten.
Vervolgens laten we het isotoop gedurende zekere tijden2.2 incorporeren waarna we de cellen lyseren door english110 ![]() l
lysebuffer (PBS-buffer pH english7.4 (bijlage
A.2english), 5% DOC (Na-deoxycholaat)
en 10% Triton X-100) toe te voegen. Daarna vriezen we de cellen in
bij -80^C.
l
lysebuffer (PBS-buffer pH english7.4 (bijlage
A.2english), 5% DOC (Na-deoxycholaat)
en 10% Triton X-100) toe te voegen. Daarna vriezen we de cellen in
bij -80^C.
Vloeibare scintillatietelling (zie fig 2.1) is
de meest gevoelige techniek voor de detectie en de meting van de radioactiviteit
in een monster. Voor de metingen kan men zowel
![]() -stralers
als electronenvangende radionucliden gebruiken [42].
In het scintillatieproces kan men verschillende stappen onderscheiden:
-stralers
als electronenvangende radionucliden gebruiken [42].
In het scintillatieproces kan men verschillende stappen onderscheiden:
Dit proces gebeurt met een maximale efficiëntie d.w.z. met een minimaal verlies aan energie. Om de electrische signalen te versterken, wordt aan het monster een scintillatiecoctail toegevoegd. Deze bestaat uit een solvent (tolueen, xyleen,..), een emulgator en een fluorescerend scintillatormolecule [21].
Problemen die bij deze techniek kunnen opduiken zijn enerzijds interferentieproblemen zoals chemoluminescentie of fotoluminescentie en anderzijds quenching (= het fenomeen dat tot een lagere scintillatietelling leidt) [21].
Men brengt het gewenste volume monster in een telflesje en voegt er 3 ml scintillatiecoctail (Optiphase HiSafe 3) aan toe. De telflesjes worden gesloten en geschud. Daarna worden ze in de vloeibare scintillatieteller (model Wallac 1409) geplaatst en wordt het programma gestart. Wij gebruiken de programma's waarbij men 1 of 3 minuten kan tellen met of zonder het afdrukken van een grafiek. In deze thesis werden enkel de grafieken van de '3 minuten'-tellingen opgenomen.
Met deze techniek verwijderen we de overmaat aan niet-geïncorporeerd
radioactief materiaal door de semipermeabele membraan van een dialysecassette.
Dit is nodig omdat het ![]() S L-methionine slechts in beperkte
mate wordt opgenomen aangezien 90-95% van dat isotoop in het medium
terechtkomt. Als we het niet zouden verwijderen, zou dit tot een hoge
radioactieve achtergrond leiden. We gebruiken Slide-A-Lyser
S L-methionine slechts in beperkte
mate wordt opgenomen aangezien 90-95% van dat isotoop in het medium
terechtkomt. Als we het niet zouden verwijderen, zou dit tot een hoge
radioactieve achtergrond leiden. We gebruiken Slide-A-Lyser![]() dialysecassettes (Pierce), die bestaan uit 2 semipermeabele membranen
omgeven door een plastic ``raamwerkje'' waarbij op de hoekpunten
ervan telkens één genummerd gaatje terug te vinden is. We gebruiken
cassettes met een cutoff van 10 000 Da, waarbij het niet-geïncorporeerde
radioactief materiaal verwijderd wordt, maar de grotere radioactief
gemerkte virusdeeltjes in de cassette achterblijven.
dialysecassettes (Pierce), die bestaan uit 2 semipermeabele membranen
omgeven door een plastic ``raamwerkje'' waarbij op de hoekpunten
ervan telkens één genummerd gaatje terug te vinden is. We gebruiken
cassettes met een cutoff van 10 000 Da, waarbij het niet-geïncorporeerde
radioactief materiaal verwijderd wordt, maar de grotere radioactief
gemerkte virusdeeltjes in de cassette achterblijven.
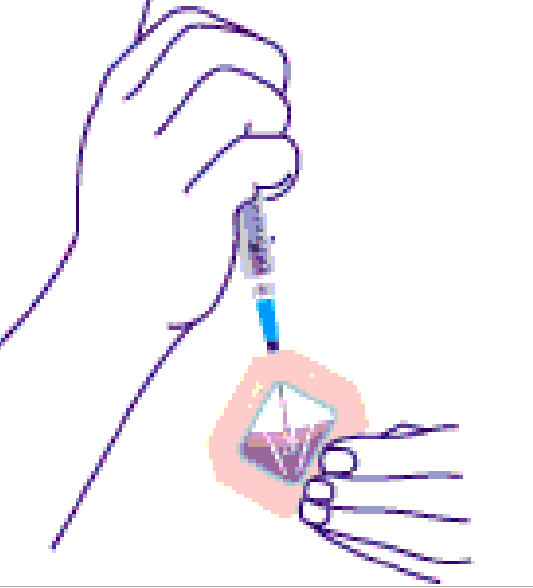
Elk monster, afkomstig van de petrischalen, werd verzameld in een apart Eppendorf cupje met de vermelding van het monster erop. Op elke dialysecassette brengt men hetzelfde merkteken aan als dat van het overeenkomstige Eppendorf cupje. Dan zuigt men m.b.v. een steriele naald en spuit de virale suspensie op. T.h.v. één van de 4 genummerde gaatjes doorprikt men voorzichtig het silicone deel binnenin de cassette totdat men de naald tussen de 2 membranen ziet verschijnen. Men spuit het virale materiaal in de cassette. Dit wordt getoond in figuur 2.2A.
Door de naald wat terug te trekken en de zuiger weer omhoog te halen,
zuigt men de lucht uit de cassette zodat de vloeistof volledig tussen
de twee membranen verdeeld is. De cassettes worden nu op een grote
vlotter bevestigd die samen met een magneetje in een recipiënt met
ongeveer 1 à 1.5 liter dialyse-buffer (zie bijlage A)
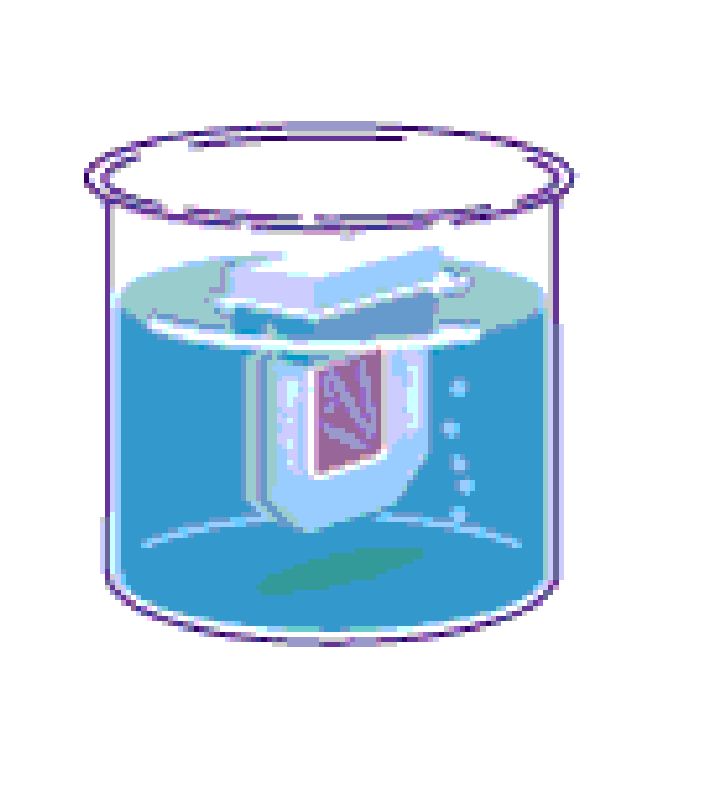
terechtkomt (zie figuur 2.2B). Het recipiënt wordt
op een magnetische roerder geplaatst zodat een vloeistofstroom ontstaat
die de dialyse bevordert. Het dialyseproces gebeurt bij 4^C, om de
degradatie van het monster t.g.v. de activatie van proteasen tegen
te gaan. We vervangen de dialysebuffer in cycli van 1.5 uur zodanig
dat de concentratiegradiënt behouden blijft en overtollig radioactief
materiaal weggespoeld wordt. Telkens we de dialysebuffer vervangen,
nemen we hiervan een monster van 500 ![]() l en bepalen we de radioactiviteit
ervan met de vloeibare scintillatieteller. We blijven dialyseren totdat
de radioactiviteit tot een aanvaardbaar niveau van 300 à 400 c.p.m.
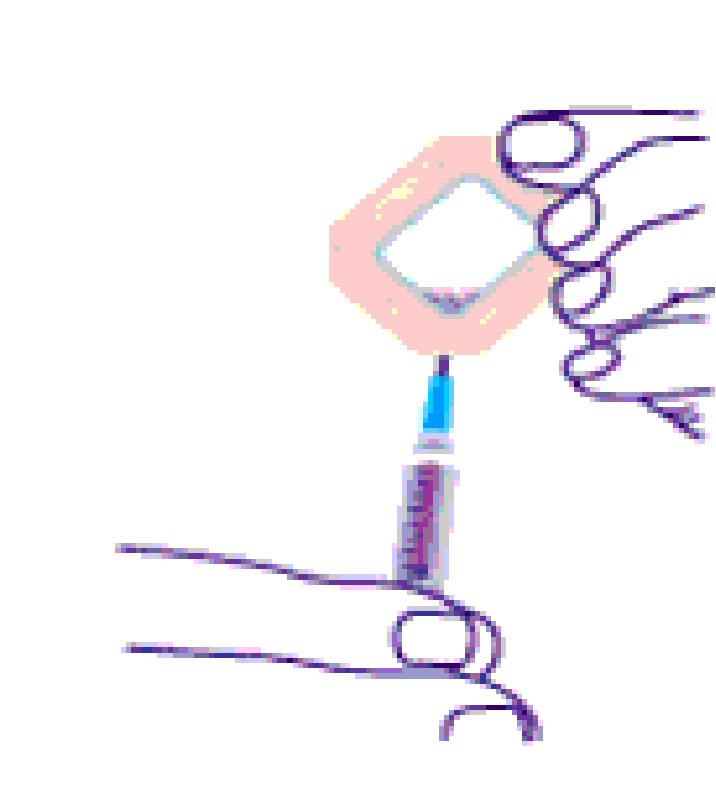
gedaald is. Dan nemen we de dialysecassette en spuiten met een steriele
naald en spuit lucht tussen de twee membranen, zodat de vloeistof
eruit kan worden gezogen zoals in figuur 2.2C.
Daarna verzamelen we het viraal materiaal in een Eppendorf cupje en
tellen we 10
l en bepalen we de radioactiviteit
ervan met de vloeibare scintillatieteller. We blijven dialyseren totdat
de radioactiviteit tot een aanvaardbaar niveau van 300 à 400 c.p.m.
gedaald is. Dan nemen we de dialysecassette en spuiten met een steriele
naald en spuit lucht tussen de twee membranen, zodat de vloeistof
eruit kan worden gezogen zoals in figuur 2.2C.
Daarna verzamelen we het viraal materiaal in een Eppendorf cupje en
tellen we 10 ![]() l van elk monster via scintillatietelling. We
centrifugeren het monster gedurende 5 minuten bij 15 000 t.p.m. (32xG)
in een eppendorfcentrifuge en bepalen tenslotte het volume van het
monster.
l van elk monster via scintillatietelling. We
centrifugeren het monster gedurende 5 minuten bij 15 000 t.p.m. (32xG)
in een eppendorfcentrifuge en bepalen tenslotte het volume van het
monster.
Bij sucrose gradiënt ultracentrifugatie (SGU) maakt men gebruik van hoge snelheden om partikels met een verschil in sedimentatiecoëfficiënt te scheiden. Daarbij brengt men het monster aan op een lineaire gradiënt van sucrose. Hierbij zullen zwaardere partikels sneller naar de bodem van de centrifugatiebuis migreren dan lichtere partikels. Op welke plaats het partikel zich uiteindelijk bevindt, is afhankelijk van de snelheid en de tijd van centrifugatie, de wrijvingscoëfficënt van het partikel, het moleculair gewicht van het partikel, de samenstelling van het omringende milieu en de afstand van de centrifugatiebuis tot het centrum van de rotor (zie sectie 2.6.2). Het gevaar bij dit soort centrifugatie bestaat erin dat als men te lang centrifugeert, alle partikels de bodem van de centrifugatiebuis bereikt zullen hebben, zodat men ze niet meer kan scheiden. De centrifugatiecondities die we gebruikt hebben zullen telkens opnieuw vermeld worden in sectie 3.
We leggen een lineaire gradiënt van sucrose (bereid in PBS-buffer; pH 7.4) aan in een centrifugatiebuis m.b.v. het toestel Watson Marlow (Model 501). Dit bestaat uit 2 buizen A en B (zie figuur 2.3) die met elkaar verbonden zijn, zodat het principe van communicerende vaten hierop van toepassing is. Onderaan buis B vertrekt -vanaf een kraantje- een slangetje dat zich opsplitst in 6 vertakkingen, die elk op de bodem van een centrifugatiebuis uitmonden. Voor de aanmaak van de 5-30% sucrose gradiënt brengen we een 30%-ige sucrose oplossing in buis A en een 5%-ige oplossing in buis B. We draaien de kraan open zodat de 5%-ige vloeistof -die rechtstreeks in verbinding staat met de slang- zo in de centrifugatiebuizen loopt. Er treedt t.h.v. de buizen A en B een drukverandering op die gecompenseerd wordt door vloeistof die vanuit buis A naar buis B stroomt. Op die manier wordt de gradiënt langzaam opgebouwd. De vloeistof in de centrifugatiebuis wordt omhooggedrukt door de nieuwe vloeistof met een hoger percentage aan sucrose, zodat men uiteindelijk de 5% sucrose bovenaan de buis terugvindt en de 30% beneden. We plaatsen de centrifugatiebuizen nu gedurende 30 minuten bij 4^C.
Bij ultracentrifugatie maken we gebruik van een Centrikon T 1170 centrifuge
en verschillende centrifugatiecondities. Voor een 15-30% gradiënt
centrifugeren we 2 uur bij 39 000 t.p.m. (200 000xG) bij een temperatuur
van 4^C, voor de scheiding van virale partikels met een sedimentatiecoëfficiënt
groter dan 20S; voor een 5-30% gradiënt is dat 17 uur aan 35 000
t.p.m. (165 000xG) bij 4^C, voor de scheiding van virale partikels
met een sedimentatiecoëfficiënt kleiner of gelijk aan 20S. Na de centrifugatie
wordt elk monster in fracties van 400 ![]() l uitgepompt m.b.v. de
Isco Density Gradient Fractioner (Model 640) in Eppendorf cupjes voor
verdere analyse of rechtstreeks in telflesjes voor scintillatietelling.
l uitgepompt m.b.v. de
Isco Density Gradient Fractioner (Model 640) in Eppendorf cupjes voor
verdere analyse of rechtstreeks in telflesjes voor scintillatietelling.
Monoclonaal antilichaam (MAb), specifiek voor een bepaald proteïne (hier anti-VP1) wordt aan een monster toegevoegd. In dit monster zal het MAb het viraal antigeen binden. De interactie tussen het virale antigeen en het antilichaam is zeer specifiek zodat enkel het viraal materiaal gebonden wordt. Dit Ab bestaat uit de Fab en de Fc fragmenten. Het Fc deel van het Ab heeft een grote affiniteit voor het oppervlakteproteïne, proteïne A, van S. aureus (Cowan I-stam). Op die manier wordt een complex (antigeen, antilichaam en bacterie) gevormd. De bacterie S. aureus wordt gebruikt omdat het een bron van proteïne A is en omdat het voor 'gewicht' bij het centrifugeren zorgt. Door centrifugatie kan het viraal materiaal van het cellulair materiaal gescheiden worden t.g.v. de sedimentatie van het antigeen/antilichaam/bacterie complex.
De monsters die we aan immunoprecipitatie onderwerpen, brengen we
op ijs, we vortexen ze en brengen van elk monster 80 ![]() l in een
afsluitbaar Eppendorf cupje. We voegen 10
l in een
afsluitbaar Eppendorf cupje. We voegen 10 ![]() l 1:50 verdund MAb
toe en laten de monsters 1 uur op het ijs incuberen zodat het antigeen
aan het antilichaam kan binden. Hierna voegen we de bacteriële suspensie
(S. aureus van de Cowan-stam) toe, en laten het geheel nog eens 30
minuten incuberen op ijs opdat het complex (antigeen/antilichaam/bacterie)
gevormd kan worden. Daarna voeren we een centrifugatiestap uit in
een Sigma 112 (B. Braun Biotech International) bij 15 000 t.p.m.
(32xG) gedurende 5 minuten. Zo scheiden we het cellulair van het viraal
materiaal. We nemen dan 80
l 1:50 verdund MAb
toe en laten de monsters 1 uur op het ijs incuberen zodat het antigeen
aan het antilichaam kan binden. Hierna voegen we de bacteriële suspensie
(S. aureus van de Cowan-stam) toe, en laten het geheel nog eens 30
minuten incuberen op ijs opdat het complex (antigeen/antilichaam/bacterie)
gevormd kan worden. Daarna voeren we een centrifugatiestap uit in
een Sigma 112 (B. Braun Biotech International) bij 15 000 t.p.m.
(32xG) gedurende 5 minuten. Zo scheiden we het cellulair van het viraal
materiaal. We nemen dan 80 ![]() l van het supernatans en brengen
dit in telflesjes voor een vloeibare scintillatietelling (zie sectie 2.4).
De rest van het supernatans wordt afgepipetteerd en het pellet wordt
geresuspendeerd in 500
l van het supernatans en brengen
dit in telflesjes voor een vloeibare scintillatietelling (zie sectie 2.4).
De rest van het supernatans wordt afgepipetteerd en het pellet wordt
geresuspendeerd in 500 ![]() l NET-Triton X-100 buffer (bijlage A.2).
Er volgt een centrifugatiestap van 5 min waarna het supernatans verwijderd
wordt (men werkt ondertussen niet meer op ijs). Het pellet wordt geresuspendeerd
in 140
l NET-Triton X-100 buffer (bijlage A.2).
Er volgt een centrifugatiestap van 5 min waarna het supernatans verwijderd
wordt (men werkt ondertussen niet meer op ijs). Het pellet wordt geresuspendeerd
in 140 ![]() l 2% SDS en ondergaat dan een kookbeurt van 5 min.
Tenslotte centrifugeert men 5 min (om het bacterieel materiaal te
verwijderen) en neemt 70
l 2% SDS en ondergaat dan een kookbeurt van 5 min.
Tenslotte centrifugeert men 5 min (om het bacterieel materiaal te
verwijderen) en neemt 70 ![]() l van het supernatans voor vloeibare
scintillatietelling. De rest wordt bij kamertemperatuur bewaard en
eventueel aan SDS-PAGE (zie sectie 2.8) onderworpen.
l van het supernatans voor vloeibare
scintillatietelling. De rest wordt bij kamertemperatuur bewaard en
eventueel aan SDS-PAGE (zie sectie 2.8) onderworpen.
SDS-PAGE of sodium dodecylsulfaat polyacrylamide gelelectroforese wordt gebruikt om de samenstelling van proteïnen te bepalen [34].
Bij deze electroforese bewegen de geladen deeltjes o.i.v. een electrisch veld van de ene pool naar de andere. Hierbij komt hun bewegingssnelheid met hun moleculair gewicht overeen. De deeltjes migreren in een gel die onstaat door polymerisatie van acrylamide (AA) en NN' methyleenbisacrylamide (BAA) waarbij TEMED (N,N,N'N' tetramethylethyleendiamine) als catalysator optreedt. Vrije radicalen geproduceerd door ammoniumpersulfaat zorgen voor de initiatie van deze polymerisatie. De poriëngrootte van de gel bepaalt hoe snel de deeltjes kunnen bewegen en deze kan men aanpassen door de concentraties van AA en BAA te wijzigen [40].
De SDS-PAGE bestaat eigenlijk uit 2 gelcompartimenten: een stapelingsgel en een scheidingsgel. De stapelingsgel bevat grote poriën en gebruikt men opdat alle monsters op dezelfde hoogte zouden starten. De poriën van de scheidingsgel zijn wat kleiner t.g.v. een hogere AA-concentratie en zorgen voor de scheiding van de gedegradeerde proteïnen.
Bij de gel voegt men vaak ook additieven zoals reducerende agentia
[![]() -Mercaptoethanol (100 mM) of Dithiothreitol (20 mM)]
of detergenten [bv: Triton X-100]. In dit geval brengen we het
proteïnenmengsel met Sodium Dodecyl Sulfaat (SDS) in contact. Dit
anionisch detergens zorgt zowel voor de degradatie van de proteïnen
als voor een netto negatieve lading in het mengsel door binding van
deze gedegradeerde proteïnen. Hierdoor kunnen ze niet meer volgens
lading gescheiden worden, maar wel volgens moleculair gewicht. Door
een electrisch veld aan te leggen zullen de negatief geladen proteïnen
door de gel migreren naar de positieve pool (de anode). Dit gaat sneller
voor proteïnen met een kleiner moleculair gewicht zodat deze lichtere
proteïnen dieper in de gel migreren dan de zwaardere.
-Mercaptoethanol (100 mM) of Dithiothreitol (20 mM)]
of detergenten [bv: Triton X-100]. In dit geval brengen we het
proteïnenmengsel met Sodium Dodecyl Sulfaat (SDS) in contact. Dit
anionisch detergens zorgt zowel voor de degradatie van de proteïnen
als voor een netto negatieve lading in het mengsel door binding van
deze gedegradeerde proteïnen. Hierdoor kunnen ze niet meer volgens
lading gescheiden worden, maar wel volgens moleculair gewicht. Door
een electrisch veld aan te leggen zullen de negatief geladen proteïnen
door de gel migreren naar de positieve pool (de anode). Dit gaat sneller
voor proteïnen met een kleiner moleculair gewicht zodat deze lichtere
proteïnen dieper in de gel migreren dan de zwaardere.
|
Vervolgens maken we de stapelings- en scheidingsgel klaar (zie tabel 2.1).
TEMED wordt als laatste component toegevoegd omdat deze de catalysator
van de reactie is, die door ![]() wordt tegengewerkt. Daarna nemen
we glazen platen die we met klemmen m.b.v. een ``latje'' op 0.5 cm
van elkaar vastklemmen. Langs de 2 zijkanten en de onderkant wordt
een laagje agar (0.5%) gegoten zodat de gel niet kan weglopen. Men
laat dit laagje even drogen. Daarna giet men de separatiegel tussen
de platen en de kam (voor de slots) wordt ook aangebracht. Na een
paar uur brengt men wat butanol op de meniscus van de scheidingsgel
zodat die overal op dezelfde hoogte komt te staan. Men spoelt na met
water en droogt met een filterpapiertje. Dan brengt men de stapelingsgel
bovenop de scheidingsgel m.b.v. een pipet. Het geheel laten we een
nacht rusten.
wordt tegengewerkt. Daarna nemen
we glazen platen die we met klemmen m.b.v. een ``latje'' op 0.5 cm
van elkaar vastklemmen. Langs de 2 zijkanten en de onderkant wordt
een laagje agar (0.5%) gegoten zodat de gel niet kan weglopen. Men
laat dit laagje even drogen. Daarna giet men de separatiegel tussen
de platen en de kam (voor de slots) wordt ook aangebracht. Na een
paar uur brengt men wat butanol op de meniscus van de scheidingsgel
zodat die overal op dezelfde hoogte komt te staan. Men spoelt na met
water en droogt met een filterpapiertje. Dan brengt men de stapelingsgel
bovenop de scheidingsgel m.b.v. een pipet. Het geheel laten we een
nacht rusten.
De loopbuffer bevat 30 g tris, 144 g glycine, 10 g SDS, aangevuld
met gedestilleerd ![]() tot 10 liter. We gebruiken een groot volume
van deze buffer omdat het voor de afkoeling van het electroforesetoestel
en de gel zorgt en omdat pH-schommelingen in een groter volume niet
zo drastisch optreden. De monsters worden in de slots van de gel gepipetteerd.
Ze lopen ongeveer 3 uur bij 70 mA/gel.
tot 10 liter. We gebruiken een groot volume
van deze buffer omdat het voor de afkoeling van het electroforesetoestel
en de gel zorgt en omdat pH-schommelingen in een groter volume niet
zo drastisch optreden. De monsters worden in de slots van de gel gepipetteerd.
Ze lopen ongeveer 3 uur bij 70 mA/gel.
Na electroforese wordt de gel 2 keer gedurende 10 minuten gewassen
met spoelvloeistof (900 ml methanol, 900 ml ![]() , 200 ml ijsazijn).
De gel blijft dan nog een nacht in deze vloeistof liggen. Daarna leggen
we de gel gedurende 15 minuten in Amplify (Amersham) zodat
de omzetting van radioactieve straling naar licht kan plaatsvinden.
Tenslotte wordt de gel gedurende 1.5 uur gedroogd en met een fotografische
film (Kodak Biomax MR-1) in contact gebracht. Afhankelijk van hoe
sterk radioactief de monsters zijn, blijft de gel een aantal dagen
(ongeveer 14 dagen) bij -80^C liggen. De ontwikkeling van de film
gebeurt in een donkere kamer: de fotografische plaat wordt ongeveer
5 minuten in een 1:5 verdunde ontwikkelingsvloeistof (LX24 van Kodak)
gelegd en vervolgens gespoeld met
, 200 ml ijsazijn).
De gel blijft dan nog een nacht in deze vloeistof liggen. Daarna leggen
we de gel gedurende 15 minuten in Amplify (Amersham) zodat
de omzetting van radioactieve straling naar licht kan plaatsvinden.
Tenslotte wordt de gel gedurende 1.5 uur gedroogd en met een fotografische
film (Kodak Biomax MR-1) in contact gebracht. Afhankelijk van hoe
sterk radioactief de monsters zijn, blijft de gel een aantal dagen
(ongeveer 14 dagen) bij -80^C liggen. De ontwikkeling van de film
gebeurt in een donkere kamer: de fotografische plaat wordt ongeveer
5 minuten in een 1:5 verdunde ontwikkelingsvloeistof (LX24 van Kodak)
gelegd en vervolgens gespoeld met ![]() . Tenslotte fixeren we de
film gedurende 5 minuten in een 1:5 verdunde fixeeroplossing (AL4
van Kodak) waarna deze opnieuw met water gespoeld wordt.
. Tenslotte fixeren we de
film gedurende 5 minuten in een 1:5 verdunde fixeeroplossing (AL4
van Kodak) waarna deze opnieuw met water gespoeld wordt.
![\includegraphics[%
width=12cm,
height=3cm]{scint_process.ps}](img32.png)
![\includegraphics[%
width=6cm,
height=6cm]{gradientmaker.ps}](img33.png)