





Publications
Presentations/Lectures

This page decribes the strenghts and limits of using RNA interference in biology and is the text version of a presentation I gave about a topic given by the jury of my PhD defense (see the presentation). Since the literature on RNA interference is quite extensive, it only comprises things that caught my particular interest ..
This page addresses :
-
Introduction into RNAi
-
Where
-
What
-
Why
-
RNAi in Biology
-
Strengths
-
Limits
-
Overcoming limits
-
Conclusion
-
References

Let's first start with the basic concepts
RNAi stands for RNA interference.
This implies that there occurs some kind of interference, .. but on
whose part ? Is it RNA that interferes with another compound, it it a
certain compound that interferes with RNA or is it actually RNA that
interferes with RNA ? The aim of this section is to clear this out.
Although the major breakthrough on "RNA interference" or "Post-Trancriptional Gene Silencing" as a general mechanism for gene silencing was first reported in the worm C. elegans, the phenomenon had long been known in fungi (like Neurospora crassa) as "Quelling" and in plants as "Co-suppression" or "Post-Trancriptional Gene Silencing". After the discovery in worms, researchers became interested in exploring whether this phenomenon was preserved in flies (Drosophila melanogaster) and mammalia. And suprise, surprise, RNA interference also occurred in these species ..
|
|
|
|
|
|
|
Quelling |
PTGS Co-suppression |
PTGS RNA interference |
PTGS |
RNA interference |




So how did researchers find out about RNA interference ? The starting assumption was that the introduction of antisense RNA would inhibit translation of mRNA complementary to the introduced asRNA. This would occur by hybridization of both strands, which physically prevented access of the ribosome to the targeted mRNA and in this way passively suppressed gene expression. An alternative way to suppress gene expression comprised of the recruitment of nucleases which led to the direct destruction of mRNA.
But, when Fire and
coworkers used this
strategy to knockdown the unc-22 gene product, they found
something bizarre.. Introduction of dsRNA, as well as single-stranded
antisense RNA ànd the control sense strand could elicit gene
silencing ?! (Although the dsRNA molecule appeared far more potent
than the other two)
What was even more puzzling wa that
this specific gene silencing could spread from one tissue to the next
and even be transmitted into the germline!
The fact that this specific silencing could spread, meant that at some point, one would require some sort of amplification mechanim, because the minuscule amounts one introduces to elicit gene silencing are not enough to dilute it out over next generations of cell or new tissues .. which means then.. that it is an active proces !
Now,
how
would this work ??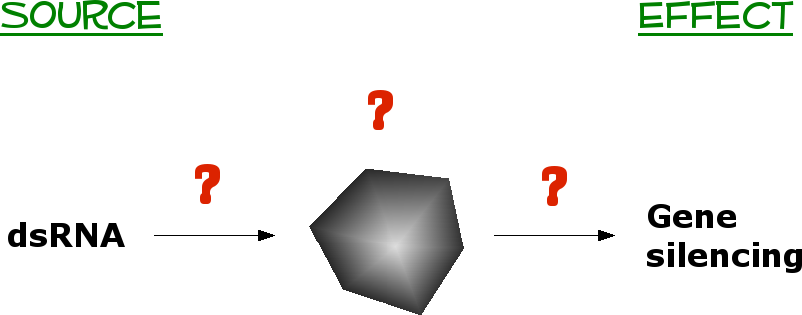
Drawn to the right, you see we have
some sort of black box scenario. This results from what we know
now : that dsRNA potently silences gene expression.
So, how can we find
out more about this
process ?
Let's take it one step at a time..
What do we know about the nature of the RNA that can induce this gene silencing ?
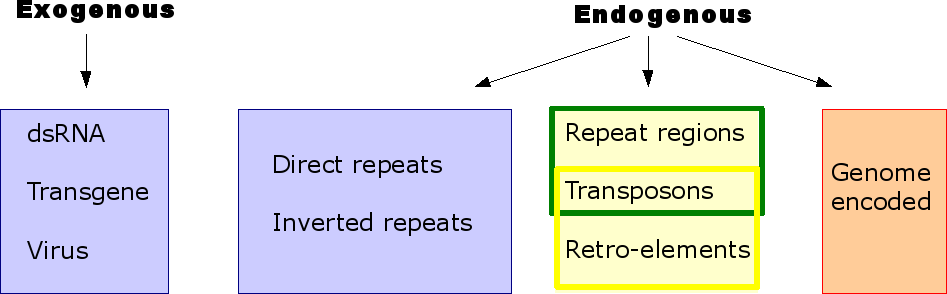 Apparantly
not all types of RNA can
induce gene silencing. Examples of RNA that can, are depicted in the
scheme on the left.
Apparantly
not all types of RNA can
induce gene silencing. Examples of RNA that can, are depicted in the
scheme on the left.
The RNA can
originate from an exogenous source like a synthetic RNA
molecule, a transgene or a virus. In addition, silencing can also be
induced
by endogenous RNA. In that case, directed or inverted repeats,
transposons and retro-elements may all be suited for the induction of
RNA interference induction (in the yellow frame endogenous RNAs that
can induce silencing in mammalia, in the green frame the ones in flies).
Now since the long dsRNA
could
obviously not be transmitted to other tissues or the next generation,
the signalling compound had to be smaller. Therefore, researchers
decided to look for small RNA molecules that occurred during PTGS in
plants. They found the small RNAs in the 20-30nt range and
characterized them into several categories depending on the origin of
their precursor. The three most well known small RNA molecules are
siRNA, miRNA and piRNA, whose origin is indicated by the blue, red and
yellow colours in the upper figure respectively. Here, "Genome encoded"
indicates precursors that originate from coding
or
noncoding transcripts that exist as individual miRNA genes or tandemly
arrayed clusters e.g. miR23.
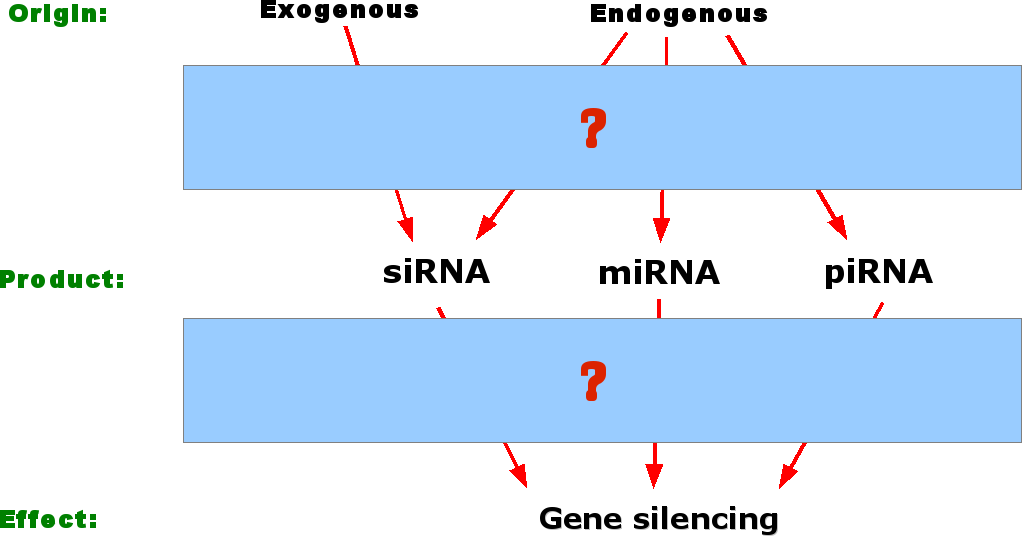
So to summarise, we
have our big RNA precursor, we
know that small
RNA molecules are involved and that this ultimately leads to gene
silencing.
So what can we
expect next ?
Reason tells us that in order to go from
large RNAs to small RNAs, we will probably need a cleavage of some
kind and to go from small RNAs to silencing of specific genes we will
probably need some homology of the small RNAs with the target genes,
that will either lead to cleavage of the targeted mRNA or prevent
translation by hybridising to the targeted mRNA.
In what follows I will mainly elaborate on the mechanims for RNA
interference or miRNA
induced silencing and refrain from discussing the piRNA induced
silencing mechanism because this is poorly characterised.
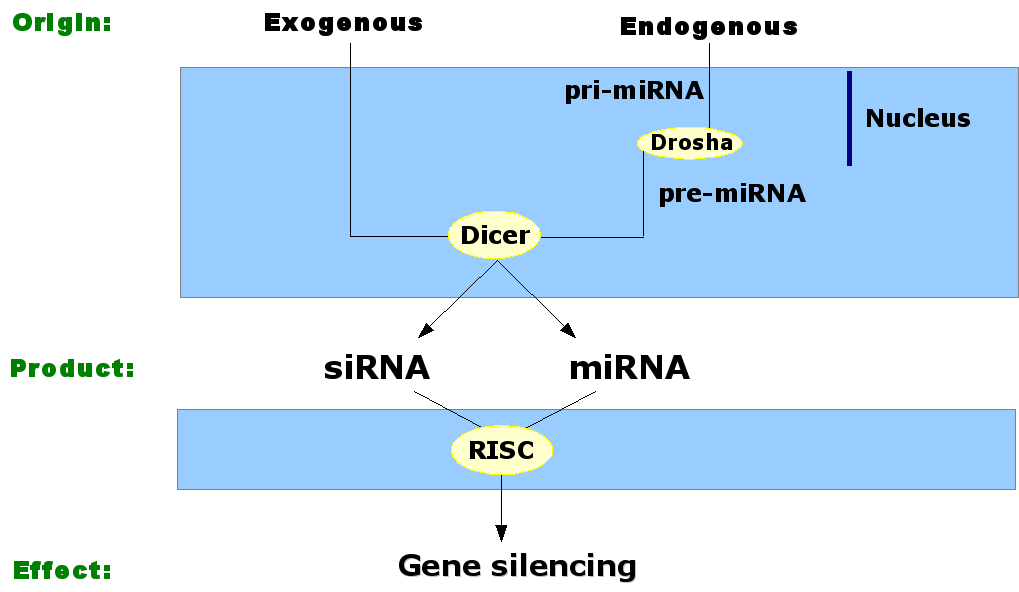 For the elucidation of the
mechanisms involved in the generation of siRNAs and miRNAs, the
breakthrough was made in plants _again_, where a nuclease called Dicer was
discovered that could produce 21-23 nucleotides with 2nt 3'
overhangs. Only one strand of the resulting dsRNA molecule will then
be incorporated into the RNA induced silencing
complex or RISC,
whereas the other strand will be degraded. The RISC complex than
finds mRNA molecules that contain complementary sequences to the
siRNA strand inside RISC and cleaves the mRNA molecule opposite to
the middle of the siRNA sequence. Although it was previously believed
that the siRNA and miRNA pathway were different pathways, nowadays
researchers believe they are the same.
For the elucidation of the
mechanisms involved in the generation of siRNAs and miRNAs, the
breakthrough was made in plants _again_, where a nuclease called Dicer was
discovered that could produce 21-23 nucleotides with 2nt 3'
overhangs. Only one strand of the resulting dsRNA molecule will then
be incorporated into the RNA induced silencing
complex or RISC,
whereas the other strand will be degraded. The RISC complex than
finds mRNA molecules that contain complementary sequences to the
siRNA strand inside RISC and cleaves the mRNA molecule opposite to
the middle of the siRNA sequence. Although it was previously believed
that the siRNA and miRNA pathway were different pathways, nowadays
researchers believe they are the same.
So therefore I will
explain
this pathway as well. Endogenous pri-miRNA transcripts are processed
by a Dicer like enzyme called Drosha, which
cleaves the precursor in
a 70nt hairpin molecule with 2nt 3' overhangs. This molecules is then
recognized by Dicer which cleaves small RNA molecules of the same
size as siRNAs, where the passenger strand of molecule is destroyed
via a bypass pathway, and the guide strand is taken up in the RISC
complex. The complex would then scan for complementary targets but
unlike in siRNA, it would lead to translational inhibition because of
some mismatches between the miRNA and the target RNA (which inhibit
binding of the ribosomes), but it would
nevertheless result in gene silencing. In plants and nematodes
silencing can spread. The protein that may help to spread the silencing
effect is called Sid2
(what Sid1 and Sid3 do, remains unknown), which can form a channel
between two adjacent cells, through which the small RNA molecule may
travel.
Why would different
organisms want to
use RNAi?
RNAi can maintain
genomic integrity by limiting the spread
of transposons and the accumulation of repititive DNA. The RNAi can
be used as a defense against viral infections. Since viruses often use
RNA intermediates during their replication process, these may be
destroyed immediately and thereby limit viral replication and spread.
Finally, miRNAs can guide gene expression. Even more they were
discovered in C. elegans to be important during development.
So let's now take a
look at how
RNA interference can be used in biology as a technique ..

In biology we use RNA interference mainly to study protein function. The advantage of using RNAi for this purpose is that we don't need to disrupt a gene about whose function we want to know more. RNAi can recognise its targets with high specificity which makes it very suitable for discriminating mutations in genes that cause diseases.
In this way, we can
compare compare
healthy and diseased cells with or without the use of siRNAs (or short
hairpin RNA molecules) or introduce these molecules at different stages
during development. We can then screen for markers of a particular
cellular process and in that way find out whether the gene of interest
may play a role there. The anwers we get from this study is that it
gives us more information about the protein and its function on the
molecular level,
but in a larger context it will enable us to unravel how signalling
is disturbed in disease processes (and thereby identify key-points for
therapeutics).
The following scheme gives a rough
indication of how we can employ RNA interference
as a method in
cell
culture:
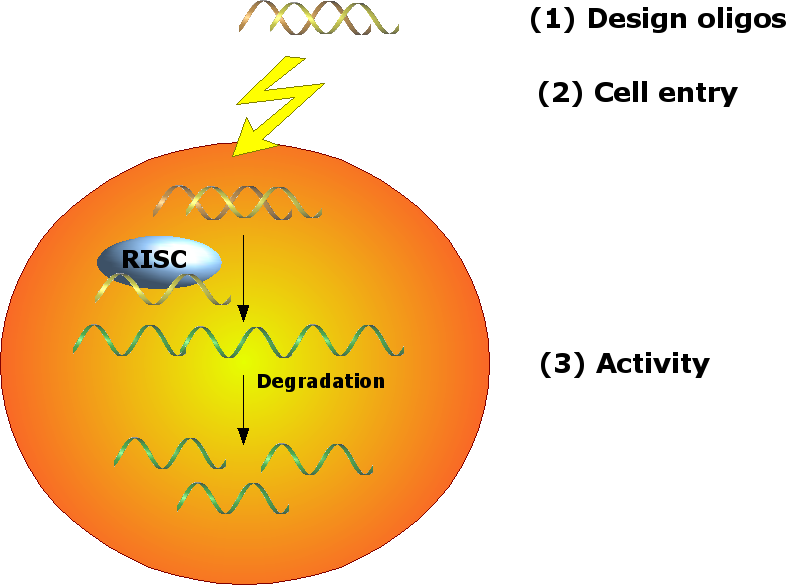 Since
we want to knockdown our “gene-of-interest”, which differs between
different research groups, we would need to devise
oligonucleotides that function as siRNA specific to the gene we want
to investigate. Once this is done, the siRNA has to get into the cell
somehow, where we later have to show that it is active, and has
silenced the gene we intended.
Since
we want to knockdown our “gene-of-interest”, which differs between
different research groups, we would need to devise
oligonucleotides that function as siRNA specific to the gene we want
to investigate. Once this is done, the siRNA has to get into the cell
somehow, where we later have to show that it is active, and has
silenced the gene we intended.
In the next section I will comment on
each step it advantages and its limits and where possible offer
solutions to
compensate for the limitations.
(1) In practice we use
siRNA molecules or
shRNA molecules. siRNA
molecules are typically 21-23 nucleotides with
2 nucleotide 3' overhangs, like they are processed by Dicer.
For
shRNAs
there exist two strategies: a type
1 hairpin has an almost
perfect match in the base and a very small loop, type 2 hairpins may
contain some mismatches and have a more extensive loop. This type of
hairpin resembles the miRNA precursor and will be dealt with as an
miRNA product by the cell.
The question for the use of these small molecules lies in how specifically such a molecule can recognize its target or rather what mismatches does it tolerate?
The first example addresses strand
specificity in viruses.
When one wants to reduce the viral spread,
RNAi could help to degrade viral RNA of particular viral genes that
are required for the propagation of the virus. For example Coxsackie
virus B3 is a positively single stranded RNA virus that belongs to
the enterovirus group in the picornaviridae. It causes myocarditis
and currently no vaccine is available. Since these viruses replicate
via the synthesis of negative ss RNA, the question is whether silencing
of genes on the positive strand ends up more advantageous for
the host cell compared to silencing of the negative strand?
In
order to solve
this question,
Schuber and collegues devised two sets of reporter vectors : one
containing the gene for EGFP, the other for luciferase. Therein, they
cloned the viral RdRP of CoxsackieB3 virus in sense and antisense
orientation and transfected this into COS7 cells together with siRNAs
against RdRP. Since siRNA molecules can recogize both the antisense
and sense RdRP gene, and the researchers wanted to be able to
discriminate between the strands they introduced Locked Nucleic Acid
(LNA)
at discrete positions: LNAs
on
the last base pair at 5'end of the sense siRNA increases its
thermodynamic stability, which
turns the 5' of the antisense into a lower binding energy pair to
ensure incorporation into RISC. Incorporation of LNA at position 10, 12
and 14 of
the sense strand impairs its participation in mRNA cleavage thereby
reducing off-target effects due to the sense strand. (The
modified strand is not
incorporated into RISC and hence will not cause silencing of of RdRP,
and inhibition of viral propagation.) By performing plaque assays, they
found out that when they targeted the positive ss viral RNA, they
required 10 to 100 times less siRNA, compared to the negative strand,
in order to obtain the same amount of viral
inhibition.
Aside from being able to discriminate
between different strands, siRNAs can apparantly also distinguish
between a few nucleotides. In a second example on the specificity
of siRNA/shRNA, researchers assessed
whether they could silence genes that differed by one nucleotide. In
this case, they wanted to distinguish between wild-type and mutated
superoxide dismutase 1 (which differ by only a single nucleotide), the
mutated form of which results in increased
toxicity during amyotrophic lateral sclerosis.
They introduced
different types of mismatches at
different position in the siRNA and assessed whether the position alone
could lead to
different extents of gene silencing. They found that mutations in all
positions, except
for position 16 would be indiscriminative. In addition to the position,
the nature of the mutation also seems to exert an effect on the
specificity of silencing. In the table below, we see the one nucleotide
difference between the WT and the disease causing mutant.
| Gene |
WT |
Mutant |
Molecule |
| SOD1 |
G |
C |
mRNA |
| C |
G |
anti-sense
siRNA molecule |
For an siRNA molecule to be able to recognize the WT or the mutant, the
complementary bases are indicated as well. Now, what seems to be so
special is that only when the mismatch between the WT molecule and the
antisense molecule that targets the mutant exists as a purine:purine
mismatch (like here G in the WT and G in the as siRNA strand), can the
siRNA discriminate between the WT and mutant. In other words, only in
these circumtances can the siRNA distinguish the mutant from the WT and
thereby destroy the mutant and preserve the WT.
These examples
obviously illustrates
the power of RNA interference to silence the target genes.
But now we wonder : do highly specific siRNAs always recognize their target with the same precision, in other words, could it be possible that once in a while unintended genes become silenced ?
In order to find this out, Jackson and coworkers designed 16 siRNAs against the same IGF1R gene and 8 siRNAs against p38® gene, also known as MAPK14. Next, they transfected HeLa cells with these siRNAs and assessed by microarray whether all siRNAs targeted the same genes as they were supposed to and whether siRNA treatment could also silence gene expression of noninteded targets. As we would expect, they found that each siRNA elicited gene silencing in the same way in three independent experiments, however, unexpectedly, they found only limited similarity in the gene expression profiles of different siRNA molecules that all targeted the same gene!
They next wondered
whether these
effects were less present at lower concentrations and thus performed
a concentration and kinetics analysis. But unlike they expected, they
could not get rid of the silencing of unintended targets. This
indicates that the concentration of siRNA is not a major factor in
off-target effects. Next, they examined the nature of the
different
transcripts that were altered by siRNA treatment and found 4 groups:
- Group 1 transcripts contained the target transcript p38® ,
- Group 3 and 4 transcripts were altered as a secondary effect due to the knockdown of p38,
- Group 2 on the other hand contained transcripts that showed homology to the siRNA molecule but were not a part of the p38 signaling pathway. Furthermore these genes were knocked down much sooner than p38, which indicates that they are probably not the result of p38 knockdown but rather due to unintended recognition of the siRNA molecules. These 9 transcripts showed homology to the core region of the siRNA or to the last 9 nucleotides from the 3' end, but only the 3' end nucleotides seemed to be involved in sequence specificity and hence were labelled as unintended off-targets.
Further analysis revealed that the minimum requirement for off-target effects with sequence homology was 11 nucleotides and that position 15 of the siRNA was the most discriminative (similar to what other groups reported later). The authors also found they could not predict which siRNA would elicit which off-targets, making this an especially nasty limit of siRNAs.
What can
we do to avoid off-target
effects when we want to investigate protein function after knockdown
of a particular gene ?
- At the design stage we can make sure that we
include the possibility for off-target effects, by searching for
homology to other sequences
at particular positions. Iyer and
coworkers developed a stand-alone program that allows for particular
requirements during the search for an effective siRNA.
- In addition,
we can also introduce LNA or other types of modifications in the sense
or antisense strand of the oligonucleotide.
- We can also use multiple siRNAs and assess each phenotype carefully. When we find a consensus phenotype for most siRNAs, it will be very unlikely that the effect can be attributed to off-target effects.
- We could compare our phenotypes with knockout cell lines.
- Finally we can try to rescue the obtained phenotype by introducing the target gene, but one that is resistant to the siRNAs
A workable vector for such a rescue approach
has been developed by Hulzel and coworkers. Here, they deigned two
episomal
constructs that
could induce
gene expression from a bidirectional promoter:
- One construct bore a luciferase and an miRNA-flanked siRNA that would target the gene of interest and thereby induce its knock-down.
- The other construct would express either the gene of interest as a rescue for the knocked down gene or a mutated gene (for example in a protein binding domain or activity domain, in order to investigate the impact of such a mutation on the function of the protein), which would need to be resistant to the siRNA. In addition, this construct also expressed a fluorescent protein as a control for the presence of the plasmid.
Both constructs
were responsive to doxcycyline, where doxcycline turned on the gene
expression and the expression of the siRNA,
whereas removal of doxycycline inhibited gene expression.
They used
rRNA processing factors PES1 and WPR12 as genes-of-interest. These
genes are involved in rRNA biogenesis and result in impaired
processing of 32S pre-rRNA and reduced cell proliferation in H1299
cells (NSCLC cells), a phenotype that can be detected easily.
Assessment of a panel of functional and
non-functional mutants of WDR12 confirmed that the assay was
functional and in addition it provided the isolation of new proteins
involved in rRNA biogenesis. Therefore, the KD-KI approach may help
to determine in a more controlled way whether a particular phenotype
is due to off-target effects or to knockdown of the targeted gene.
(2) Since we now have an idea about what factors to take into account when designing oligos, we can proceed to the next step, namely the introduction of siRNAs in the cell. This part also includes a brief mention on the responses of the cell to the introduction of this foreign material.
Plants and worms
are able
to take up siRNAs from their environment and can transmit this siRNA
signal to other cells or distribute it over their
tissues and even transmit it to the next generation. This contrasts to
mammalian cells that unable to spontaneously take up siRNAs. This means
that we need an
artifical way to introduce these compounds into the cell. We can do
this with invasive techniques like micro-injection and
electroporation, or we can deliver siRNAs in a more natural way : via
liposomes, via cell penetrating peptides or internalizable
antibodies. The fact that the receptivity of each cell type towards
these molecules determines the efficiency of silencing, means
that silencing effect may very between transfected cells.
In contrast to
plants and worms, mammalia lack the amplification and propagation
responses. This implies that
when we introduce siRNAs into mammalian cells the RNAi effect only
lasts temporarily and in order to overcome this caveat, we may
introduce
shRNA via viral vectors or the construction of stable cell lines
expressing the shRNA.
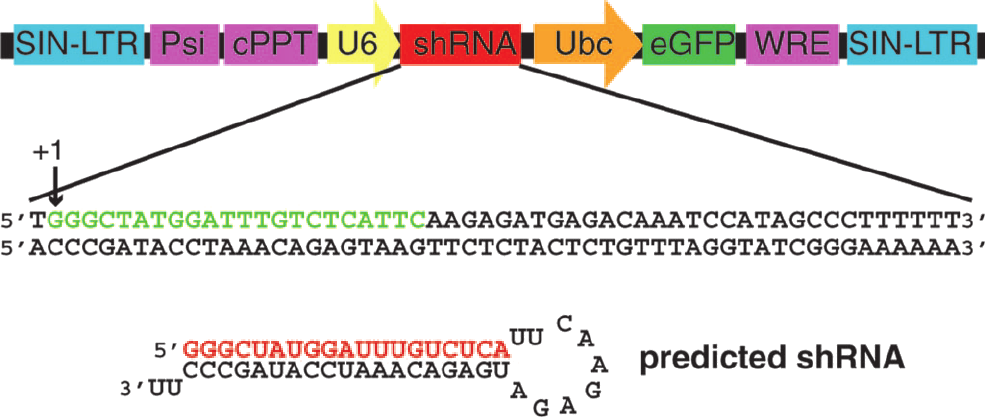
An example of such a methodology was
recently provided by Dann and coworkers. They knocked down the gene for
Dazl,
an RNA-binding protein required for fertility. In mice, knock-out of Dazl results in male and female
sterility. When using the shRNA vector (indicated in the figure on the
left), they found male sterility, which nicely indicates how RNAi can
indicate the involvement of a particular
gene product in physiological function.
Figure from PNAS
2006;103(30):11246. Dann et al.
However, the introduction of siRNA is a
quite stressfull event for the cell, and numerous reports have
described that shRNA and siRNA can elicit the interferon stress
response in cells, which ultimately leads to apoptosis or cell
death.
This reaction is vertebrate-specific and is initiated when dsRNA, blunt
ended ds or ss siRNAs are introduced into the cell or when the cell can
detect 2'OH uridines. In contrast to the off-targeting effects, this
response is concentration-dependent,
and occurs even when the molecule shows no similarity to
the
gene-of-interest.
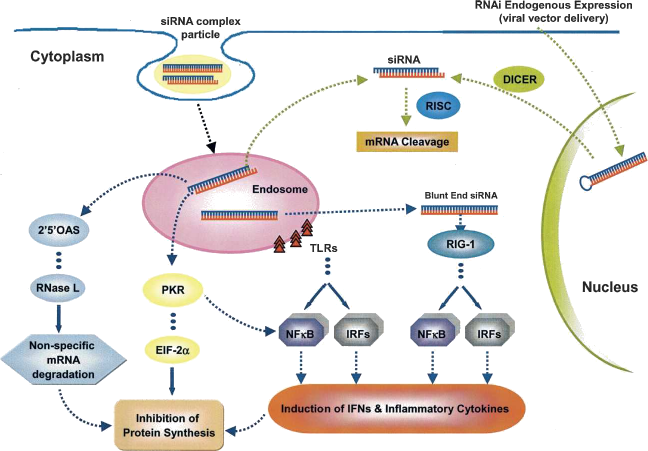 The cellular
stress response
The cellular
stress response
The figure on the right (from "RNA 2007;13:431. De Paula et al.") provides an overview of the different stress pathways that can all lead to inhibition of protein synthesis and consequently cell death upond the introduction of foreign material. The most famous pathway is the PKR pathway where dsRNA activates PKR which then phosphorylates eIF-2alpha which then leads to inhibition of protein synthesis. dsRNA can also trigger oligoadenylate synthases which then activates RNAseL and causes unspecific degradation of mRNA. Blunt ended siRNA can be recognized by Retinoic-acid Inducible Gene 1(RIG-1) which unwinds the molecule and thereby activate NF·B and Interferon Responsive Factor 3 (IRF). These compounds induce the synthesis of inflammatory cytokines and thereby the death of the cell. Similarly Toll-like receptors (TLRs) in the endosomal vesicle, can also induce NF·B and IRFs.
How can we avoid triggering the stress
response of the cell?
For siRNAs, we can
:
- avoid A/U nucleotides at the 5' end,
- design siRNAs so that they won't hybridize with sites for mRNA-binding proteins
- assure that :
- the molecule contains 2nt 3' ends for more effective cleavage
- hybridization between the siRNA and the target mRNA results in an A-form helix.
- the G/C content lies between36-56%
- the siRNA has the most effective size : 21-23nucleotides or 25-29 nucleotides.
(3) When we have introduced our siRNA into the cell, we can next concern ourselves with the activity of the RNAi machinery. Besides the introduction of a single siRNA or shRNA one can also knock-down multiple genes at once. So let's see what this possibility has in store for us.
This approach may help us target genes that encode redundant proteins at once, an approach considered by Zhu and colleagues. They constructed a vector with a replacable promoter and two miRNA flanking sites (for long-term expression and transcription from a Pol II promoter) in between which one can insert a shRNA.
In a first stage, they tested whether they could downregulate 3 genes in single, double and triple KD constructs for Arrestin2, GPCR kinase 2 and G-protein beta-2; and in a next step they created stable cell lines of RAW macrophages to KD gene pairs of Arrestin 2/3, Grk2/5 and PKAcalpha/beta. In these cell lines they could KD the proteins as expected.
Since they
previously performed a
microarray experiment to screen for cAMP dependent mRNAs, they
selected four genes for assessment in the PKAc®
/¯ KD cell
line.These genes were :
- Nr4a1 and Nr4a2,which are members of the nuclear orphan receptors regulated by cAMP;
- Ctla2® and Ctla2¯,
which are
cysteine protease inhibitors originally isolated in cytotoxic
T-lymphocytes, whose transcriptional regulation has not been
investigated
in this context.
KD of either PKAc® or PKAc¯ did not result in a
decreased response after administration of cAMP analogues, however
double KD markedly decreased the transcripts of these four genes. This
indicates that KD of redundant genes may indeed provide a clearer
picture of a proteins function or involvement.
After being able to target redundant
proteins at once, the question is whether one can KD genes that
signal in the same pathways to estimate the role of these genes
in a
particular processes targeted by the pathway. For this Sahin and
coworkers knocked down the ErbB2 receptor tyrosine kinase and the
protein
kinases Akt and MEK1. They first determined at which concentrations
all three genes would be moste effectively downregulated without
causing too much cytotoxicity. This turned out to be the lowest
concentration. Next, they assessed whether other genes that
would not signal in the same pathway were affected. They found that
although RNA levels could display high fluctuations, these were not
translated onto the protein level when all three genes were knocked
down.
To determine whether the knock-down of different combinations of these gene would result in differences in cellular function, they transfected cells with, single, double and triple KD shRNAs and assessed their invasion potential. Their results confirm that Akt, one of the main players during metatasis, is mainly involved in invasion and the ERK1/2 pathway in proliferation. ErbB2 KD does not reduce the invasion capacity and suggests that Akt can be activated via other means. A double KD of ErbB2 and MEK1 implies that other receports can target Akt which leads to a reduced cell invasion. The triple KD does not completely eliminate invasion potential thereby suggesting that parallele mechanisms can intervene to attenuate the invasion potential.
Although off-targeting remains a problem in these experiments, things are even more complicated with multiple siRNAs or shRNAs: RISC can only bind a limited amount of siRNAs, so the use of high concentrations of siRNAs results in many unbound molecules in the cytoplasm and the initiation of the cellular stress response. Even more, during increased siRNA concenrations, only the “fittest” siRNAs are incorporated into RISC leaving the less potent molecules wandering in the cytoplasm. shRNAs can cause saturation because they compete with the endogenous miRNA pathway for processing power. Such competition can induce toxicity as the next example illustrates.
Grimm and coworkers introduced either liver-targeted luciferase or human ®1 anti-trypsin and specific shRNA in mice. This introduction caused weight loss and death when high concentrations were administered. They found that this toxicity was accompanied by a reduction in the amount of liver miR-122 and let7a and remained independent of liver injury induced by chemicals or surgery. shRNA function was decreased in the presence of shRNA or miRNAs and indicated that shRNA indeed competes with the miRNA machinery. One bottleneck for processing seems to be exportin 5, the nuclear exporter of pre-miRNA, since overexpression of this protein, increased silencing again in presence and absence of shRNA/miRNA competitors.
The last example of how siRNAs or shRNAs may be used is at an even more increased level of complication, namely the genome-wide RNA interference. Such an approach may yield a lot of data and understanding but it requires specialized equipment(robotics) and very carefull planning.
An example of such a genome-wide analysis concerns a cell morphology study where two different Drosophila cell lines where treated with a dsRNA library and microscopically checked for parameters like cell number, size, shape, viability, actin filaments, microtubules and DNA. In addition, a co-RNAi screen was performed with PTEN to assess its role in cell shape. Knock-down of approximately 100 genes resulted in a visible effect amongst which several modifiers for PTEN, a phosphatase that dephosphorylates PIP3 (and a functional antagonist of PI3K).
This indicates that when performed carefully, genome-wide RNAi screens may yield valuable data on protein function and their roles in cellular processes, providing that all information is carefully validated. Unfortunately, today there exists too little overlap (stochastic and biological errors,off-target effects..) between the results obtained in different genome wide RNAi studies to incorporate all data in the same databases.
Conclusion :
Using RNA
interference in biology provide very usefull. However, despite it
strenghts (specificity, multiple targets and use in transgenic
animals), its use also includes several limitations (off-target
effects, cellular stress responses, saturation.
Ideally, one can -at
least- partially account for off-targets by carefull design of oligos.
By including at least one marker for the interferon stress response it
should be possible to discern whether or not one elicits a such a
response. In addition, one can use several siRNAs against the same
target gene, to partially determine which genes are unwelcomed targets
and to determine phenotypes, it may help to use a knock-down rescue
model to ensure that the phenotype one sees does not result from
off-target effects, saturation effects or stress responses.
References
:
Current Opinion in Molecular Therapeutics;2003:5(3):217 Paddison and
Hannon.
Current Opinion in Molecular Therapeutics;2006:9(3):248. Svoboda.
Nature Reviews Genetics 2007;8:884. Chapman et al.
Nature 1998;391(6669):806. Fire et al.
Nature 2004;431:338. Mello and Conte.
Nature 2004;431:343.Meister and Tuschl.
Nature 2004;431:371. Hannon and Rossi.
Nature 2004;431:364.Lippman and Martienssen.
Nature 2004;431:350. Ambros.
Antiviral Research 2007;73:197. Schubert et al.
PloS Genetics 2006;2(9):e140. Schwarz et al.
Nature Biotechnology 2003;21(6):635. Jackson et al.
RNA 2007;13:431. De Paula et al.
Cold Spring Harbor Symposia on Quantitative Biology LXXL, 2007. Prevot
and Perrimon.
BMC Molecular Biology 2007,8:98. Zhu et al.
PNAS 2007;104(16):6579. Sahin et al.
PNAS 2006;103(30):11246. Dann et al.
Nucleic Acids Research 2007;35(3):e17. Hülzel et al.
J.Biology 2003;2:27. Kiger et al.
Computer Methods and Programs in Biomedicine 2007;85:203. Iyer et al.
Nature 2006;441:537. Grimm et al.
Nucleic Acids Research 2005;33(1):439. Elmen et al.
PNAS 2006;103(15):5953. Dykxhoorn et al.
RNA 2006;12:1197. Jackson et al
Published 06-12-2007 on Yellowcouch.org