





Publications
Presentations/Lectures
The roles of mammalian mitogen-activated protein kinase-activating protein kinases (MAPKAPKs) in cell cycle control
Summary
1. The cell cycle
The cell cycle in somatic cells of multicellular organisms is a complex process that consists of four successive stages: the G1, S, G2 and M phase (Figure 1). During the G1 (gap) phase, the cell prepares for DNA replication, while DNA replication itself occurs in the S (synthesis) phase. After the S phase, cells are stalled in a second gap phase (G2), during which the replicated DNA is checked for mistakes and, if necessary repaired before it is passed on to the daughter cells. The replicated chromosomes are separated during the M (mitosis) phase, after which two daughter cells arise. These can, in turn re-enter the cell cycle [reviewed in Morgan, 2007]. In gametocytes, the mitosis phase is replaced by meiosis, which can be subdivided in meiosis I and meiosis II [Marston and Amon, 2004; Pawlowski and Cande, 2005; van den Heuvel, 2005].Quiescent cells that no longer divide enter a so-called G0 phase [reviewed in Morgan, 2007].
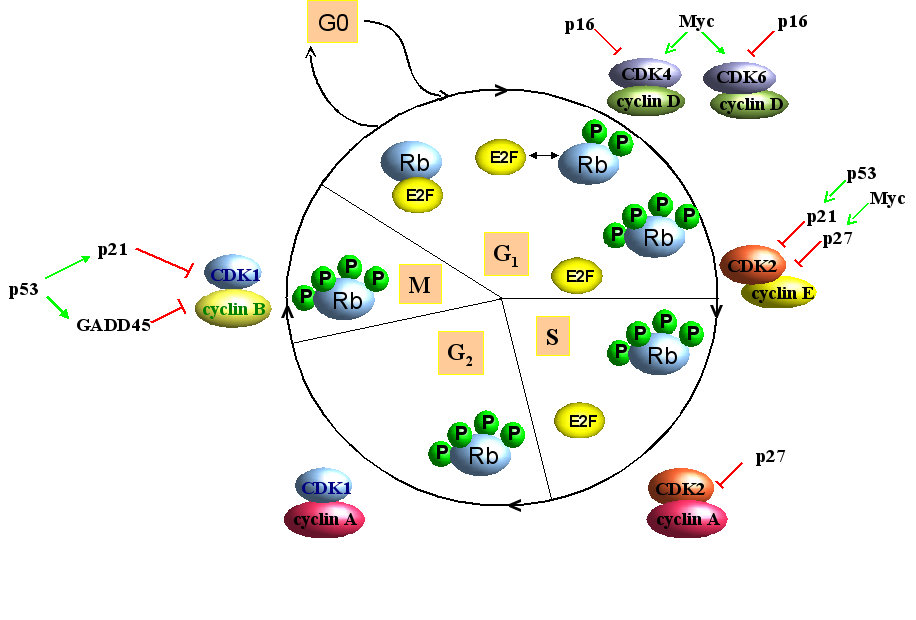
Figure 1. Simplified presentation of the cell cycle in somatic cells. The cell cycle can be divided in 4 successive phases: G1, S, M and G2. Non-dividing cells enter the G0 phase. Complexes of cyclin/Cyclin-dependent kinases (Cdk) promote cell progression, while Cdk inhibitors p16, p21 and p27 counteract the action of cyclin/Cdk complexes. Unphosphorylated retinoblastoma (Rb) protein prevents G1/S transition by hampering the activity of E2F, while hyperphosphorylation inhibits this action of Rb. Other proteins such has the tumor suppressor p53 and the oncogene Myc are also implicated in cell cycle regulation (see text for details).
2. The mitogen-activated protein kinase (MAPK) pathway
As the name suggests, mitogens were the first group of signals identified that stimulated these pathways, but also stress and cytokines can elicit activation of the MAPK pathways. MAPK pathways regulate cellular processes such as proliferation, survival/apoptosis, differentiation, development, adherence, motility, metabolism, and gene regulation. The classical mitogen-activated protein kinase (MAPK) signaling pathways consist of cascade of three consecutive phosphorylation events exerted by a MAPK kinase kinase (MAPKKK), a MAPK kinase (MAPKK), and a MAPK. MAPK in turn phosphorylates non-protein kinase substrates or yet other protein kinases. The latter are referred to as MAPK-activated protein kinases (MAPKAPK). Figure 2 illustrates the different MAPK signaling modules identified in mammalian cells [Johnson and Lapadat, 2002; Roux and Blenis, 2004; Imajo et al., 2006; Cuevas et al., 2007; Raman et al., 2007; Weston et al., 2007; Zhang et al., 2007].
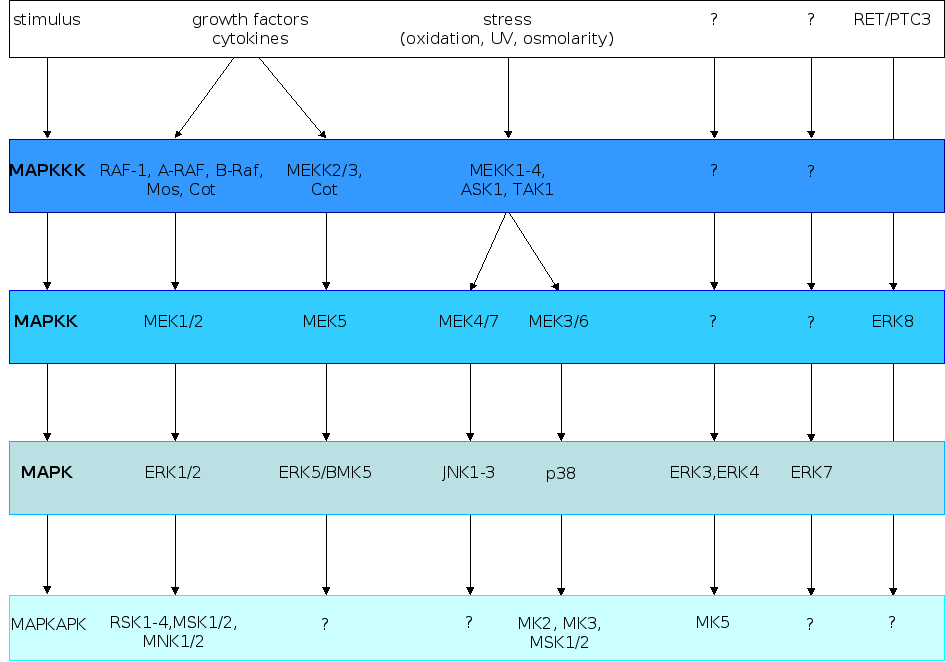
Figure 2. The different
MAPK cascades
in mammalian cells. The typical MAPK pathway consists of a tripartite
module in which a MAPKKK phosphorylates a MAPKK, which in turn
phosphorylates a MAPK. The natures of the signals that activate the
different MAPK cascades are shown at the top of the figure, while
some of the substrates are indicated at the bottom of the figure. The
poorly identified atypical ERK3, ERK4, ERK7 and ERK8 pathways are
also depicted. The figure is adapted from [Imajo
et al., 2006].
3. The role of MAPKAPK in cell cycle regulation
3.1. p90 ribosomal S6 kinase or RSK
3.1.1. Properties and function of RSK
RSK was originally detected in Xenopus laevis oocytes as a 90 kDa protein kinase that phosphorylated the 40S ribosomal subunit protein S6. RSK is also known as p90Rsk or MAPKAPK1 because this serine/threonine kinase acts downstream of the MAPK ERK1/2 (Figure 2). RSKs are present in most vertebrate species, as well as in several invertebrates examined [Dai et al., 2008 and references therein]. Two RSK isoforms (RSK1 and RSK2) have been identified in Xenopus, while four mammalian RSK members (RSK1-4) have been described. Though similar in structure to other family members, RSK4 differs in its function from RSK1-3 by the presence of a unique region through which it can inhibit the MAPK signaling pathway. This inhibitory role is dependent on a region that is not conserved in RSK1-3 [Myers et al., 2004]. RSKs are involved in several cellular functions and regulate: i. gene expression by modulating the activity of transcription factors and chromatin remodeling factors; ii. protein syntheses by phosphorylation of polyribosomal proteins; iii. signal transduction pathways such as PKA, NFB, Toll-like receptor signaling through cross-talk; iv. cell survival by targeting proteins involved in apoptosis; v. differentiation; and vi. cell cycle [Frödin and Gammeltoft, 1999; Hauge and Frödin, 2006; Zaru et al., 2008; Gerits et al. 2007a].
3.1.2. The role of RSK in cell cycle regulation
The four isoforms for RSK described in mouse and human (RSK1-4) seem all implicated in cell cycle regulation, but their precise role and the exact mechanisms by which they affect the cell cycle is poorly understood. In the first part of this section, we will review examples of studies that suggest the involvement of RSK isoforms in cell cycle regulation. Then we will elucidate the role and mechanism of RSK1 in cell cycle regulation in oocytes. Finally, we present other mechanisms by which RSK may modulate the cell cycle.
General observations that suggest a role for RSK in cell cycle regulation
RSK1 was reported to be involved in G1/S transition because the specific inhibitor of RSK1, SL0101 (see section 4.), produced a block in the G1 phase of the cell cycle in MCF-7 cell. Similar effects on cell cycle progression were obtained after depletion of RSK1 [Smith et al., 2005]. Although the molecular mechanism remains unclear, these data indicate that in order to pass the G1 restriction point, these cells require RSK1 activity. The RSK2 isoform may also play a role in cell cycle progression because RSK2-/- mouse embryonic fibroblasts accumulate at the G1 phase, while RNA interference showed that knockdown of RSK2 expression arrested the cells in G1 [Smith et al., 2005; Cho et al., 2007]. Depletion of RSK4 strongly reduced the mRNA levels of p21Cip1 and abolished p53-dependent G1 cell arrest induced either by conditional activation by p53 or by DNA damage via ionizing irradiation [Berns et al., 2004]. These observations underscore a role for RSK4 in cell cycle regulation, but the mechanism of action of RSK4 in these processes was not addressed.
The role of the cytostatic factor (CSF) and the anaphase-promoting complex/cyclosome (APC/C) in oocyte maturation
The best understood role of RSK in modulating the cell cycle comes from studies in oocytes of the African clawed frog Xenopus laevis. In most animals, development of immature oocytes into mature oocytes is arrested in the meiotic cell cycle. Unfertilized oocytes then await fertilization before the cell cycle will progress. This arrest occurs at the metaphase of meiosis II in vertebrates and is referred to as cytostatic factor (CSF), where CSF does not describe a single protein, but rather a cell division inhibitory activity in the oocyte. Maturation of oocytes requires the activity of a factor that was originally termed maturation-promoting factor (MPF). MPF is now known to be a universal complex, composed of cyclin B and Cdk1 that drives mitosis and meiosis in all eukaryotic cells. The enzymatic activity of MPF (or cyclin B/Cdk1) changes throughout oocyte maturation, but could be detected in the M-phase from yeast to man. CSF holds the anaphase-promoting complex/cyclosome (APC/C/C) in an active state. APC/C is a mitosis-specific E3 ubiquitin ligase that can induce degradation of cyclin B to promote anaphase entry. Inhibition of APC/C will therefore prevent degradation of cyclin B and this will contribute to activation of cyclinB/Cdk1 (MPF). Thus CSF, which ensures metaphase arrest, can via APC/C affect the activity of MPF, which promotes metaphase→anaphase progression (see Figure 3). RSK, as outlined below, controls the activity of both CSF and MPF.
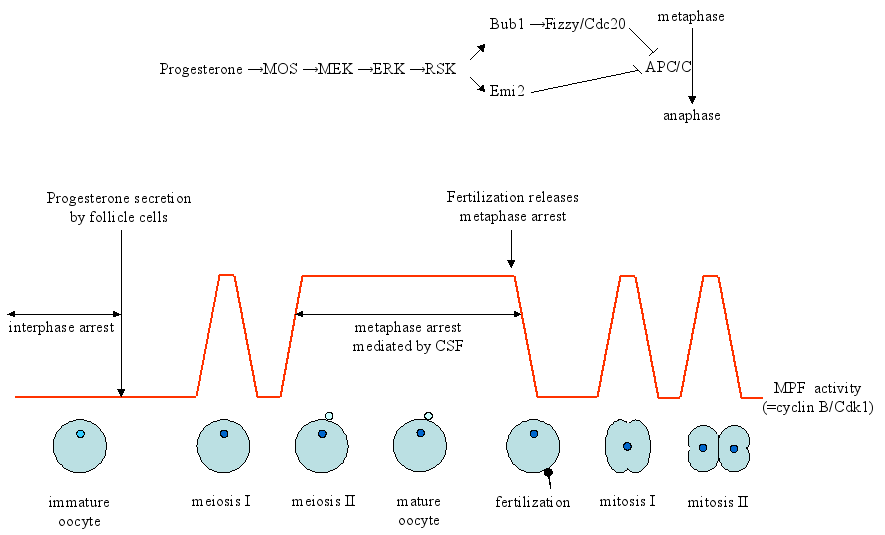
Figure 3. Pathways involved in maturation of Xenopus laevis oocyte. Immature oocytes are arrested at the border of the first meiotic division (meiosis I). Progesterone induces maturation of the oocytes, allowing them to enter meiosis I and subsequently meiosis II. The oocytes accumulate thereafter in metaphase due to a cell division inhibitory activity referred to as cytostatic factor (CSF). Fertilization of the egg overcomes CSF arrest and allows exit from meiosis II and entry into embryonic division of the fertilized egg. Two pathways contribute to establishment and maintenance of CSF, including cyclinB/Cdk1 (=MPF; maturation-promoting factor) and the MOS-MEK1-ERK-RSK pathway. The anaphase promoting complex/cyclosome (APC/C) governs the levels of cyclin B. In immature oocytes, cyclinB/Cdk1 activity is low because of inhibitory phosphorylation of threonine-14 and tyrosine-15. Progesterone leads to the synthesis of MOS and activation of the MAP kinase cascade MEK1-ERK-RSK. RSK can phosphorylate Emi2, which releases Emi2 from APC/C and activates APC/C. RSK also phosphorylates and activates Bub1, which prevents Cdc20 to activate the APC/C complex. See text for details. The figure is adapted from [Tunquist and Maller, 2003].
The effect of RSK on the spindle check point component Bub1
One mechanism by which RSK exerts its effect on the cell cycle relies on phosphorylation of Bub1 (budding uninhibited by benzimidazole) protein kinase. This modification leads to inhibition of the anaphase-promoting complex (APC/C). The activity of APC/C is controlled by the co-activators Fizzy/Cdc20 and Cdh1. Fizzy/Cdc20 activates APC/C at the metaphase to anaphase transition, whereas Cdh 1 functions during late mitosis and early G1 phase. Phosphorylation of Bub1 by RSK prevents binding of Fizzy/Cdc20 to APC/C. As a result APC/C is inhibited and this blocks the metaphase/anaphase transition and causes CSF arrest [Schwab et al., 2001; Tunquist et al., 2002; for reviews see Maller et al., 2002; Tunquist and Maller, 2003; Liu et at., 2007]. The spindle assembly/kinetochore attachment checkpoint, required for correct alignment of the chromosomes, forms also restriction point that keeps cells in the metaphase. Once the chromosomes are properly attached, this arrest is relieved and cells enter the anaphase, during which the chromosomes are equally segregated between the two daughter cells. The spindle assembly checkpoint is composed of multiple proteins, including Mad, Bub and Mps. Hence, modulation of the function of spindle checkpoint components (e.g. by RSK-mediated phosphorylation of Bub) can block anaphase progression [reviewed in Tunquist and Maller, 2003 and in Morgan, 2007].
RSK and the APC/C inhibitor early mitotic inhibitor 2 (Emi2)
Another target for RSK is the zinc finger-containing protein Emi2 (early mitotic inhibitor 2; also known as Erp1). During CSF arrest, Emi2 is bound to APC/C and inhibits APC/C function. Upon fertilization, calmodulin-dependent kinase II phosphorylates Emi2, which results in rapid Emi2 degradation. Cdk1 can also phosphorylate Emi2 and this modification disrupts the binding of Emi2 to APC/C. Depletion of Emi2 or dissociation of Emi2 from APC/C alleviates APC/C inhibition, and promotes M phase exit into the first embryonic interphase. RSK can also phosphorylate Emi2, thereby enhancing Emi2’s stability and activity [Nishiyama et al., 2007; Inoue et al., 2007]. In this regard, RSK-mediated phosphorylation of Emi2 at Ser-335 and Thr-336 stimulated the interaction with the protein phosphatase PP2A, resulting in dephosphorylation of Cdk1 phosphorylation sites on Emi2. This increases the stability of Emi2, which maintains CSF arrest through inhibition of APC/C [Wu et al., 2007]. Gross and co-workers also observed a decrease in inhibitory phosphorylation of Cdk1 at Tyr-15 upon microinjection of constitutive RSK1 in Xenopus oocytes, but the exact mechanism was not examined [Gross et al., 2001]. A possible mechanism for RSK-induced dephosphorylation of Cdk1 at Tyr-15 was suggested by the findings of Chen and Gardner and is outlined in Figure 4. They found that RSK was also implicated in endothelin-triggered G2/M progression of vascular smooth muscle cells. Thus, endothelin activates the MEK/ERK pathway which results in the activation of RSK. Active RSK phosphorylates Myt1, which inactivates the enzymatic activity of Myt. Hence, Myt-mediated inhibitory phosphorylation of Cdk1 subsides and Cdk1 becomes activated [Chen and Gardner, 2004]. In another study, an alternative route for RSK2-Emi1-APC/C was identified. The authors showed that RSK2 can phosphorylate the APC/C inhibitor Emi1. This phosphorylation stimulated complex formation between phospho-Emi1 and the APC/C activator Cdc20. Thus in this scenario, Emi1 phosphorylated by RSK2 sequesters Cdc20 from APC and helps to establish CSF arrest during mouse oocyte maturation [Paronetto et al., 2004].
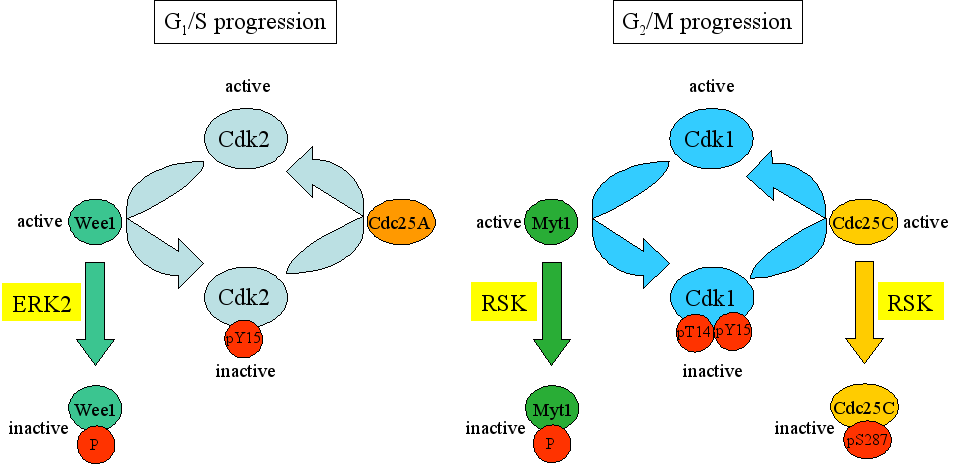
Figure 4. Phosphorylation and dephosphorylation events of Cdk2 and Cdk1 regulate G1/S and G2/M progression, respectively. The Wee1 kinase inactivates Cdk2 through inhibitory phosphorylation. Wee1 itself is inactivated through ERK2-mediated phosphorylation. The phosphatase Cdc25A can remove this inhibitory phosphorylation, converting Cdk2 into an active state. Similarly, Myt1 inactivates Cdk1 through phosphorylation. This inactivation can be counteracted by Cdc25C which dephosphorylates Cdk1 and thereby renders it active. The activities of Myt1 and Cdc2C are controlled by RSK.
RSK and cyclin B
Another mechanism by which RSK may modulate cell cycle progression in oocytes is through stimulation of cyclin B synthesis [Gross et al., 2001]. The molecular mechanism by which RSK stimulates synthesis of cyclin B remains unsolved.
RSK and the Cdk activator Cdc25
One way by which ERK2 may contribute towards G2 arrest is by phosphorylating and activating Wee1, the protein kinase that catalyzes the inhibitory phosphorylation of Cdk1 (Figure 4, left panel). An additional route to prevent G2 exit is through ERK2-induced activation of RSK and subsequent phosphorylation Cdc25C. This will also affect G2/M progression (Figure 4, right panel). It was demonstrated that ERK2 activates RSK, which in turn can phosphorylate Cdc25C at Ser-287. Phosphorylated Cdc25C binds 14-3-3 and keeps Cdc25C in an inactivate state, or/and stimulates nuclear exclusion. As a result, Cdk1 does not become dephosphorylated and the cyclinB-Cdk1 complex is retained inactive. Thus RSK-mediated phosphorylation of Cdc25C at Ser-287 contributes to G2 arrest in Xenopus oocytes [Chun et al., 2005]. The ERK2-RSK pathway may also be used to keep other cell types in the G2 phase. Indeed, treatment of human cervical cancer HeLa cells with epidermal growth factor or with the phorbol ester TPA resulted in delayed M-phase entry and coincided with phosphorylation of and reduction in activity of Cdc25C. Both epidermal growth factor and TPA can activate ERK2, but the involvement of RSK was, however, not tested [Barth et al., 1996].
The role of RSK in oocyte maturation of other species
RSK is not only involved in oocyte maturation of mammals and Xenopus, but also of other species. PhosphoRSK protein levels were differently expressed during developmental stages of Artemia parthenogenetica (brine shrimps), and RSK activation was coupled to termination of G2/M arrest. Moreover, in vivo reduction of RSK activity by either RNA interference, the specific pharmacological inhibitor SL0101, or antibody neutralization with anti-phospho-RSK antibodies resulted in inhibition of mitosis in the cells of Artemia embryos, suggesting a role for RSK in termination of the G2/M arrest and promotion of mitogenesis during the post-embryonic development of Artemia-encysted embryos, although the details remain obscure [Dai et al., 2008]. However, the involvement of RSK in regulation of the cell cycle in oocytes seems to be species specific. While RSK1 and RSK2 contribute to Xenopus laevis oocyte arrest in metaphase of the second meiotic division [Bhatt and Ferrell, 1999; Gross et al., 1999], the RSK isoforms 1, 2, and 3 are dispensable for this arrest in mouse oocytes [Dumont et al., 2005]. Moreover, the phase of cell cycle arrest is also species specific. While the female gametes of most animals are arrested in the meiotic cell cycle awaiting fertilization, the cell cycle in oocytes of starfish (Asterina pectinifera) is arrested in the G1 phase. This arrest requires RSK and inhibition of RSK (by use of an inhibitory antibody) released the arrest and initiated DNA replication without fertilization, while maintenance of RSK activity (by use of a constitutive active variant) prevented DNA replication following fertilization [Mori et al., 2006]. The exact mechanism by which RSK prevents cell cycle progression in unfertilized egg cells of the starfish remains to be solved.
Other mechanisms by which RSK may interfere with cell cycle progression
Another mechanism by which RSK influences the cell cycle is through the Cdk inhibitor p27Kip1. Both RSK1 and RSK2 were demonstrated to directly bind and phosphorylate p27Kip1 at serine-10 and threonine-198 in vivo. The phosphorylation of threonine-198, but not serine-10, promoted the interaction of p27Kip1 with the 14-3-3 isoforms and , and to a lesser degree to 14-3-3 and 14-3-3. Phosphothreonine 189 could not bind to 14-3-3 and 14-3-3. Binding of the phosphothreonine-198 p27Kip1 to 14-3-3 stimulated cytoplasmic localization of this complex. Since p27Kip1 negatively regulates G1 cell cycle progression by inactivating cyclinE-Cdk2 and cyclinA-Cdk2 complexes, RSK may promote G1/S transition by inducing nuclear exclusion of p27Kip1 [Fujita et al., 2003]. The biological relevance of RSK-mediated phosphorylation of p27Kip1 on serine-10 remains unclear.
3.2. MSK1 and MSK2
3.2.1. Properties and functions of MSK1 and MSK2
The human and mouse genome encodes two mitogen- and stress-activated protein kinases, referred to as MSK1 and MSK2. MSK1 and MSK2 are substrates for both ERK and p38 MAPK and are widely expressed (Figure 2). Studies in cells have assigned a role for MSK1/2 in regulation of translational control (through phosphorylation of the eIF4E-binding protein 4E-BP1) and in transcriptional control (through phosphorylation of transcription factors such as ATF1, CREB, ER81, c-FOS, p65 subunit of NFB, STAT3, and chromatin remodeling factors such as histone H3, HMGN1) [reviewed in Dunn et al., 2005]. The precise biological roles of MSK1 and MSK2 are incompletely understood because single and double knock out mice were viable and fertile, without any obvious phenotype. However, in contrast to control littermates, MSK1-/- mice displayed no enhanced locomotion after repeated cocaine exposure and showed lower preference for cocaine [reviewed in Gerits et al., 2007a]. Moreover, mice lacking MSK1 show impaired Pavlovian fear conditioning and spatial learning [Chwang et al., 2007].
3.2.2. The role of MSK1 and MSK2 in cell cycle regulation
MSK1 phosphorylated the protein kinase LKB1 (see section 3.1.2) at Ser-431 in vitro, but LKB1 did not seem to be a physiological substrate for MSK1. Studies by Sapkota et al. revealed that RSK mediated phosphorylation of LKB1 (see 3.1.). The fact that RSK appears in larger amounts in cells may explain why RSK rather than MSK phosphorylated LKB1 in vivo. On the other hand, stimuli that specifically activate MSK1 did not induce phosphorylation of LKB1 at Ser-431, jeopardizing a role of MSK1 as LKB1 kinase [Sapkota et al., 2001]. MSK1/2 may be indirectly involved in nerve growth factor (NGF) induced G1/S arrest during neuronal differentiation of PC12 cells. NGF treatment resulted in activation of MSK1/2 and this promoted dissociation of the PCAF-PP1/PP2A cytoplasmic complexes, coincident with PCAF phosphorylation and nuclear translocation. PCAF then mediated acetylation of p53 at lysine-320, thereby enhancing the transcriptional activity of p53. As a result, transcription of the p53 target gene encoding cyclin-dependent kinase inhibitor p21Cip1 increased and cells are arrested in G1 phase [Wong et al., 2004].
3.3. MNK1 and MNK2
3.3.1. Properties and function of MNK1 and MNK2
MNK1 and MNK2 (MAPK signal-integrating kinase) were originally identified by screening ERK substrates, but later it was shown that they are activated in vivo by both ERK and p38 MAPK. One single MNK1 protein and two isoforms of MNK2, MNK2a and MNK2b, have been identified in human and mouse. MNK1 and MNK2 can phosphorylate eIF4E (also known as cap-binding protein) at Ser-209, but the biological significance of eIF4E phosphorylation and its effect on the regulation of protein synthesis is controversial [Ueda et al., 2004]. The exact in vivo functions of MNK1 and MNK2 are not known because mice deficient in MNK1, MNK2, or both, appear normal [reviewed in Gerits et al., 2007a]. A recent study reported that transgenic mice expressing a constitutive active MNK1 mutant developed rapid tumors compared to control mice, suggesting that MNK1 can act as an oncogene [Wendel et al., 2007]. MNK1 and MNK2 seem to be involved in anti-apoptotic signaling in response to serum withdrawal as well [Chrestensen et al., 2007].
3.3.2. The role of MNK1 and MNK2 in cell cycle regulation
To our best knowledge, no direct functional link between MNK1 and MNK2 and the cell cycle has been reported so far, although it was demonstrated that overexpression of a dominant negative MNK1 inhibited proliferation of cells [Worch et al., 2004; Wendel et al., 2007]. However, MNK1 and MNK2 may indirectly modulate the cell cycle by regulating the activity of eIF4E, which affects the transport of cyclin D1 mRNA from the nucleus to the cytoplasm or upregulates translation of the mRNA for cyclin-dependent kinase inhibitor p27Kip1 [Topisirovic et al., 2002; Eto, 2006].
3.4. MK2
3.4.1. Properties and function of MK2
Three structurally related enzymes referred to as MK2, MK3/3pK, and MK5/PRAK act downstream of p38 MAPK (Figure 2), but activation by ERK1/2 and JNK has also been observed, at least in vitro. MK2 was the first of these three MAPKAPKs to be isolated and is by far the best studied. This protein kinase is expressed in all invertebrates and vertebrates examined so far, but a structural homolog in yeast is lacking [reviewed in Gaestel, 2006]. Studies with knock-out mice revealed that MK2 is essential for liposaccharide-induced cytokine biosynthesis, which is essential for the inflammatory response [Kotlyarov et al., 1999]. Consistent with this role, MK2 deficient mice show increased susceptibility to infection [Lehner et al., 2002], but the function of MK2 also extends to other cellular processes such as actin remodeling and cell migration by phosphorylation of Hsp25/27, stabilization of mRNA, chromatin remodeling and gene regulation, and regulation of the cell cycle [Heidenreich et al., 1999; Lasa et al., 2000; Janknecht, 2001; Yannoni et al., 2004; Kobayashi et al., 2006; reviewed in Gaestel, 2006].
3.4.2. The role of MK2 in cell cycle regulation
Several observations have pointed to the involvement of MK2 in cell cycle regulation. For example treatment of Swiss 3T3 cells with fibroblast growth factor-2 or hemopoietic cells with granulocyte colony-stimulating factor resulted in proliferation and coincided with strong increase in MK2 activity, suggesting that MK2 can trigger cell cycle progression [Maher, 1999; Rausch and Marshall, 1999]. Both MK2 and MK5 could inhibit Ras-induced proliferation of NIH 3T3 cells. Although the exact mechanism for this anti-mitogenic effect was not known, it was shown that MK2 and MK5 could ablate AP1- and SRE-dependent transcription by inhibiting Ras-induced JNK, but not ERK activation [Chen et al., 2000]. More recent studies have provided insight into the molecular mechanisms by which MK2 may affect the cell cycle.
3.5. MK3
3.5.1. Properties and function of MK3
MK3/3pK was detected by two independent research groups by two different approaches. The team of Rapp identified the gene encoding MK3 when they analyzed chromosome 3p21.3 in small lung cancer tumor suppressor gene region, while McLaughlin and colleagues identified MK3 when searching for interaction partners for p38 MAPK [Ludwig et al., 1996; McLaughlin et al., 1996; Sithanandam et al., 1996]. MK3 expression is restricted to birds and mammals and its activation exclusively depends on p38/ MAPK [reviewed in Gaestel, 2006]. Furthermore, MK3 regulates chromatin remodeling/transcriptional activation via polycomb, a multi-protein complex that contributes to stable silencing of specific genes and repression of the transcription factor E47 [Neufeld et al., 2000; Voncken et al., 2005].
3.5.2. The role of MK3 in cell cycle regulation
So far, no direct role for MK3 in the cell cycle has been identified. However, overexpression of MK3 resulted in de-repression of the gene encoding the p53-stabilizing protein p14/p19ARF and slowed down cell cycling [Voncken et al., 2005]. MK3-induced cell cycle arrest corresponds well with the known biological feature of ARF to induce both G1 and G2 arrest (and hence senescence; see 3.6.2.) due to its stabilizing effects on the p53 transcription factor [Gallagher et al., 2006; Sherr, 2006].
3.6. MK5
3.6.1. Properties and function of MK5
In 1998 a novel MAPKAPK was identified by two independent laboratories and named MAPKAPK-5 (MK5) or PRAK (p38-regulated and –activated kinase), respectively. PRAK is the human homolog of MK5 [New et al., 1998; Ni et al., 1998]. MK5 seems to be ubiquitously expressed in all vertebrates examined so far, but is absent in Caenorhabditis elegans and Drosophila [reviewed in Gaestel, 2006]. The biological functions of MK5 are poorly understood. To determine the in vivo role of MK5, knock out mice or mice that express a constitutive active MK5 mutant were generated. Depending on the genetic background of the mice, MK5-/- animals appear normal with no obvious phenotype (129xC57BL/6 background) or have increased embryonic lethality (C57BL/6 background) [reviewed in Gerits et al., 2007a]. MK5 deficient mice do not develop spontaneous tumors within 2 years after birth, but display increased sensitivity to chemical-induced skin cancer. Moreover the authors showed that MK5 mediated senescence upon activation by p38 MAPK in response to oncogenic p21RAS, and that MK5 deficiency rendered primary cells more susceptible to oncogenic transformation, suggesting a tumor suppressing role for MK5 [Sun et al., 2007]. Mice expressing a constitutive active MK5 demonstrated anxiety-related traits and locomotor differences compared to their control littermates, which may implicate a role for MK5 in neurological processes [Gerits et al., 2007b]. Moreover, we recently characterized an in vivo interaction between the PKA and MK5 and demonstrated that MK5 is involved in PKA-induced microfilament rearrangement [Gerits et al., 2007c].
3.6.2. The role of MK5 in cell cycle regulation
Senescence, a permanent state of cell-cycle arrest, develops naturally in most cells after repeated cell division, but can be induced prematurely by multiple DNA damage, telomere dysfunction, derepession of the INK4a/ARF locus and other stimuli. Senescence suppresses the development of cancer cells by arresting the proliferation of damaged cells that are at risk for malignant transformation [Campisi and d’Adda di Fagana, 2007; Collado et al., 2007; Yaswen and Campisi, 2007]. A previous study had demonstrated that wild-type, but not a kinase dead mutant of MK5 could prevent p21RAS-induced proliferation in NIH3T3 cells [Chen et al., 2000]. In a later studie, Sun and colleagues expanded this finding by showing that MK5 mediated activated (oncogenic) p21RAS-induced senescence and that MK5 was required for p21RAS-induced silencing of cyclin A. Moreover, MK5 phosphorylated p53 at Ser-37 in the transactivation domain and stimulated the transcriptional activity of p53 [Sun et al., 2007]. Thus both MK3 and MK5 may be involved in senescence, MK3 by ARF-mediated stabilization of p53 and MK5 by increasing the activity of p53 through direct phosphorylation at Ser-37..
One genuine substrate for MK5 is the atypical MAPK ERK3 [Schumacher et al., 2004; Seternes et al., 2004]. The interaction between ERK3 and MK5 stabilizes ERK3. It was recently shown that ERK3 directly binds cyclin D3, as well as Cdc14A (an antagonist of cyclin-dependent kinase 1), and that Cdc14A stabilizes complex formation between ERK3 and cyclin D3 [Sun et al., 2006; Hansen et al., 2007]. These findings suggest a putative role for ERK3 in the cell cycle. Because MK5 binds to and phosphorylates ERK3 and stabilizes ERK3 [reviewed in Gaestel, 2006], MK5 may through ERK3 affect the cell cycle. Moreover, overexpression of ERK3 inhibits S phase entry in mouse NIH3T3 fibroblasts. Enrichment of ERK3 in the nucleus or in the cytoplasm markedly attenuated ERK3’s ability to block S phase entry, indicating that nucleocytoplasmic shuttling of ERK3 is important [Julien et al., 2003]. We and others have previously shown that co-expression of ERK3 and MK5 results in nuclear exclusion of ERK3 [Schumacher et al., 2004; Seternes et al., 2004]. Thus MK5 could through retaining ERK3 in the cytoplasm prevent ERK3 to inhibit G1/S phase transition.
4. MAPKAPK inhibitors and therapeutic applications
The implication of MAPKAPK in cell cycle regulation makes these protein kinases attractive therapeutic targets in conditions with perturbed cell cycling. In the last years, tremendous efforts have been made to design specific MAPKAPK inhibitors. Some examples will be discussed here.
5. Conclusion
Loss of normal cell cycle regulation and uncontrolled cell proliferation, a hallmark for tumor cells, can result from perturbed action of protein kinases [Greenman et al., 2007]. The last decennium has therefore seen an explosion in the development of specific protein kinase inhibitors and their use in clinical trials [Mikalsen et al., 2006]. MAPKAPK seem to be implicated in cell cycle progression, but a putative role for all known MAPKAPK in cell cycle control has not been addressed. A challenge for future research is to scrutinize the mechanisms by which MAPKAPK can affect the cell cycle. Generating mouse models or cell lines deficient in MAPKAPK or the use of RNA interference to knock down the expression of a particular MAPKAPK may increase our knowledge of the functions of that MAPKAPK. A better understanding of the fine molecular mechanisms by which a certain MAPKAPK affects the cell cycle may allow the development of highly efficient and specific MAPKAPK inhibitors.
6. Acknowledgement
Research in our laboratory is financed by the Norwegian Cancer Society (Kreftforeningen; projects A01307).7. References
Due to the enormous number of published articles on the issue of cell cycle (>280,000 hits for the key word cell cycle on PubMed in the beginning of 2008), we had to make a stringent selection and we apologize to those whose work has not been referred to.- Abraham,R.T. (2005). MAPKAP kinase-2: three's company at the G(2) checkpoint. Molecular Cell, 17(2): 163-164.
- Alessi D.R., Sakamoto K., Bayascas J.R. (2006). LKB1-dependent signaling pathways. Annual Review of Biochemistry, 75:137-163.
- Bartek, J., Lukas J. (2003). Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell, 3(5): 421-429.
- Bartek, J., Lukas J. (2007). DNA damage checkpoints: from initiation to recovery or adaptation. Current Opinion in Cell Biology, 19(2): 238-245.
- Barth H., Hoffmann I., Klein S., Kaszkin M., Richards J., Kinzel V. (1996). Role of cdc25-C phosphatase in the immediate G2 delay induced by the exogenous factors epidermal growth factor and phorbolester. Journal of Cellular Physiology, 168(3): 589-599.
- Berns K., Hijmans E.M., Mullenders J., Brummelkamp T.R., Velds A., heimerikx M., Kerkhoven R.M., Madiredjo M., Nijkamp W., Weigelt B. Agami R., Ge W., Cavet G., Linsley P.S., Beijersbergen R.L., Bernards R. (2004). A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature, 428(6981): 431-437.
- Bhatt R.R., Ferrell J.E. Jr. (1999). The protein kinase p90 rsk as an essential mediator of cytostatic factor activity. Science, 286(5443): 1362-1365.
- Blais A., Dynlacht B.D. (2007). E2F-associated chromatin modifiers and cell cycle control. Current Opinion in Cell Biology, 19(6): 658-662.
- Bloom J., Cross F.R. (2007). Multiple levels of cyclin specificity in cell-cycle control. Nature Reviews. Molecular Cell Biology, 8(2): 149-160.
- Brown J.R., Nigh E., Lee R.J., Ye H., Thompson M.A., Saudou F., Pestell R.G., Greenberg M.E. (1998). Fos family members induce cell cycle entry by activating cyclin D1. Molecular and Cellular Biology, 18(9): 5609-5119.
- Bulavin D.V., Higashimoto Y., Popoff I.J., Gaarde W.A., Basrur V., Potapova O., Appella E., Fornace A.J., Jr. (2001). Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature, 411 (6833): 102-107.
- Campisi J., d’Adda di Fagagna F. (2007). Cellular senescence: when bad things happen to good cells. Nature Reviews. Molecular Cell Biology. 8(9): 729-740.
- Cánepa E.T., Scassa M.E., Ceruti J.M., Marazita M.C., Carcagno A.L., Sirkin P.F., Ogara M.F. (2007). INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB, 59(7): 419-426.
- Chen R. H., Abate C., Blenis J. (1993). Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. The Proceedings of the national Academy of Sciences of the United States of America, 90(23): 10952-10956.
- Chen G., Hitomi M., Han J., Stacey D.W. (2000). The p38 pathway provides negative feedback for Ras proliferative signaling. The Journal of Biological Chemistry, 275(50): 38973-38980.
- Chen S., Gardner D.G. (2004). Suppression of Wee1 and stimulation of Cdc25A correlates with endothelin-dependent proliferation of rat aortic smooth muscle cells. The Journal of Biological Chemistry, 279(14): 13755-13763.
-
- Cho Y.Y., Yao K., Kim H.G., Kang B.S., Zheng D., Bode A.M., Dong Z. (2007). Ribosomal S6 kinase 2 is a key regulator in tumor promoter induced cell transformation. Cancer Research, 67(17): 8104-8112.
- Chrestensen C.A., Shuman J.K., Eschenroeder A., Worthington M., Gram H., Sturgill T.W. (2006). MNK1 and MNK2 regulation in HER overexpressing breast cancer cell lines. Targeting the Kinome Meeting, December 4th-6th, Basel, Switzerland, Poster 15.
- Chrestensen C.A., Eschenroeder A., Ross W.G., ueda T., Watanabe-Fukunaga R., Fukunaga R., Sturgill T.W. (2007). Loss of MNK function sensitizes fibroblasts to serum-withdrawal induced apoptosis. Genes to Cells, 12(10): 1133-1140.
- Chun J., Chou A.S.S., Maingat F.G., Edmonds S.D., Ostergaard H.L., Shibuya E.K. (2005). Phosphorylation of Cdc25C by pp90Rsk contributes to a G2 cell cycle arrest in Xenopus cycling egg extracts. Cell Cycle, 4(1): 148-154.
- Chwang W.B., Arthur J.S., Schumacher A., Sweatt J.D. (2007). The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. The Journal of Neuroscience, 27(46): 12732-12742.
- Clark D.E., Errington T.M., Smith J.A., Frierson H.F., Weber M.J., Lannigan D.A. (2005). The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Research, 65(8) :3108-3016.
- Cobrinik D. (2005). Pocket proteins and cell cycle control. Oncogene, 24(17): 2796-2809.
- Collado M., Blasco M.A., Serrano M. (2007). Cellular senescence in cancer and aging. Cell, 130(2): 223-233.
- Cuevas B.D., Abell A.N., Johnson G.L. (2007). Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene, 26(22): 3159-3171.
- Dai J.Q., Zhu X.J., Liu F.Q., Xiang J.H., Nagasawa H., Yang W.J. (2008). Involvement of p90 ribosomal S6 kinase in termination of cell cycle arrest during development of Artemia-encysted embryos. The Journal of Biological Chemistry, 283 (3): 1705-1712.
- Dumont J., Umbhauer M., Rassinier P., Hanauer A., Verlhac M.H. (2005). P90Rsk is not involved in cytostatic factor arrest in mouse oocytes. The Journal of Cell Biology, 169(2): 227-231.
- Dunn K.L., Espino P.S., Drobic B., He S., Davie J.R. (2005). The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochemistry and Cell Biology, 83(1): 1-14.
- Eto I. (2006). "Nutritional and chemopreventive anti-cancer agents up-regulate expression of p27Kip1, a cyclin-dependent kinase inhibitor, in mouse JB6 epidermal and human MCF7, MDA-MB-321 and AU565 breast cancer cells". Cancer Cell International, 6:20.
- Fisher R.P. (2005). Secrets of a double agent: CDK7 in cell-cycle control and transcription. Journal of Cell Science, 118(Pt 22): 5171-5180.
- Frödin M, Gammeltoft S. (1999). Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Molecular and Cellular Endocrinology, 151(1-2): 65-77.
- Fujita N., Sato S., Tsuruo T. (2003). Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal S6 kinase promotes its binding to 14-3-3 and cytoplasmic localization. The Journal of Biological Chemistry 278(49): 49254-49260.
- Gaestel M. (2006). MAPKAP kinases - MKs - two's company, three's a crowd. Nature Reviews. Molecular Cell Biology, 7(2): 120-130.
- Gallagher S.J., Kefford R.F., Rizos H. (2006). The ARF tumour suppressor. International Journal of Biochemistry & Cell Biology, 38(10): 1637-1641.
- Gerits N., Kostenko S., Moens U. (2007a). In vivo functions of mitogen-activated protein kinases: conclusions from knock-in and knock-out mice. Transgenic Research, 16(3): 281-314.
- Gerits N., Van Belle W., Moens U. (2007b). Transgenic mice expressing constitutive active MAPKAPK5 display gender-dependent differences in exploration and activity. Behavioral and Brain Functions, 3(1): 58.
- Gerits N., Mikalsen T., Kostenko S., Shiryaev A., Johannessen M., Moens U. (2007c). Modulation of F-actin rearrangement by the cyclic AMP/cAMP-dependent protein kinase (PKA) pathway is mediated by MAPK-activated protein kinase 5 and requires PKA-induced nuclear export of MK5. The Journal of Biological Chemistry, 282(51): 37232-37243.
- Gerits N., Kostenko S., Shiryaev A., Johannessen M., Moens U. (2008). Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility.Acceptance pending.
- Giacinti C., Giordano A. (2006). RB and cell cycle progression. Oncogene, 25(38): 5220-5227.
- Giono L.E., Manfredi J.J. (2006). The p53 tumor suppressor participates in multiple cell cycle checkpoints. Journal of Cellular Physiology, 209(1): 13-20.
- Greenman C., Stephens P., Smith R., Dalgliesh G.L.,Bignell G., Davies H., Teague J., Butler A., Stevens C., Edkins S., O'Meara S., Vastrik I., Schmidt E.E., Avis T., Barthorpe S., Bhamra G., Buck G., Choudhury B., Clements J., Cole J., Dicks E., Forbes S., Gray K., Halliday K., Harrison R., Hills K., Hinton J., Jenkinson A., Jones D., Menzies A., Mironenko T., Perry J., Raine K., Richardson D., Shepherd R., Small A., Tofts C., Varian J., Webb T., West S., Widaa S., Yates A., Cahill D.P., Louis D.N., Goldstraw P., Nicholson A.G., Brasseur F., Looijenga L., Weber B.L., Chiew Y.E., DeFazio A., Greaves M.F., Green A.R., Campbell P., Birney E., Easton D.F., Chenevix-Trench G., Tan M.H., Khoo S.K., Teh B.T., Yuen S.T., Leung S.Y., Wooster R., Futreal P.A., Stratton M.R.(2007). Patterns of somatic mutation in human cancer genomes. Nature, 446(7132): 153-158.
- Gross S.D., Schwab M.S., Lewwellyb A.L., Maller J.L. (1999). Induction of metaphase arrest in cleaving Xenopus embryos by the protein kinase p90Rsk. Science, 286(5443): 1365-1367.
- Gross S.D., Schwab M.S., Taieb F.E., Lewellyn A.L., Qian Y.W., Maller J.L. (2000). The critical role of the MAP kinase pathway in meiosis II in Xenopus oocytes is mediated by p90(Rsk). Current Biology,10(8): 430-438.
- Gross S.D., Lewellyn A.L., Maller J.L. (2001). A constitutively active form of the protein kinase p90Rsk1 is sufficient to trigger the G2/M transition in Xenopus oocytes. The Journal of Biological Chemistry, 276(49): 46099-46103.
- Guay J., Lambert H., Gingras-Breton G., Lavoie J.N., Huot J., Landry J. (1997). Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. Journal of Cell Science, 110 ( Pt 3): 357-368.
- Hansen C.A., Bartek J., Jensen S. (2007). A functional link between the human cell cycle-regulatory phosphatase Cdc14A and the atypical mitogen-activated kinase Erk3. Cell Cycle, 7(3) [Epub ahead of print].
- Hauge C., Frödin M. (2006). RSK and MSK in MAP kinase signalling. Journal of Cell Science, 119(Pt 15): 3021-3023.
- Heidenreich O., Neininger A., Schratt G., Zinck R., Cahill M.A., Engel K., Kotlyarov A., Kraft R., Kostka S., Gaestel M., Nordheim A. (1999). KAP kinase 2 phosphorylates serum response factor in vitro and in vivo. The Journal of Biological Chemistry, 274(20):14434-14443.
- Huard S., Elder R.T., Liang D., Li G., Zhao R.Y. (2007). HIV-1 Vpr induces cell cycle G2 arrest through Srk1/MK2-mediated phosphorylation of Cdc25. Journal of Virology, Dec 26 [Epub ahead of print].
- Imayo M., Tsuchiya Y., Nishida E. (2006). Regulatory mechanisms and functions of MAP kinase signaling pathways. IUBMB Life, 58(5-6): 312-317.
- Inoue D., Ohe M., Kanemori Y., Nobui T., Sagata N. (2007). A direct link of the Mos-MAPK pathway to Erp1/Emi2 in meiotic arrest of Xenopus laevis eggs. Nature, 446(7139): 1100-1104.
- Janknecht R. (2001). Cell type-specific inhibition of the ETS transcription factor ER81 by mitogen-activated protein kinase-activated protein kinase 2. The Journal of Biological Chemistry, 276(45): 41856-41861.
- Johnson G.L., Lapadat R. (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science, 298(5600): 1911-1912.
- Julien C., Coulombe, P., Meloche S. (2003). Nuclear export of ERK3 by a CRM1-dependent mechanism regulates its inhibitory action on cell cycle progression. The Journal of Biological Chemistry, 278(43):42615-42624.
- Katajisto P., Vallenius T., Vaahtomeri K., Ekman N., Udd L., Tiainen M., Mäkelä T.P. (2007). The LKB1 tumor suppressor kinase in human disease. Biochimica et Biophysica Acta, 1775(1): 63-75.
- Kellogg D.R. (2003). Wee1-dependent mechanisms required for coordination of cell growth and cell division. Journal of Cell Science, 116(Pt 24): 4883-4890.
- Kobayashi M., Nishita M., Mishima T., Ohashi K., Mizuno K. (2006). MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO Journal, 25(4): 713-726.
- Kotlyarov A., Neininger A., Schubert C., Eckert R., Birchmeier C., Volk H.D., Gaestel M. (1999). MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nature Cell Biology, 1(2): 94-97.
- Lasa M., Mahtani K.R., Finch A., Brewer G., Saklatvala J., Clark A.R. (2000). Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Molecular and Cellular Biology, 20(12): 4265-4274.
- Lemaire M., Froment C., Boutros R., Mondesert O., Nebreda A.R., Monsarrat B., Ducommun B. (2006). CDC25B phosphorylation by p38 and MK-2. Cell Cycle, 5(15): 1649-1653.
- Lehner M.D., Schwoebel F., Kotlyarov A., Leist M., Gaestel M., Hartung T. (2002). Mitogen-activated protein kinase-activated protein kinase 2-deficient mice show increased susceptibility to Listeria monocytogenes infection. Journal of Immunology, 168(9): 4667-4673.
- Liu J., Grimison B., Maller J.L. (2007). New insight into metaphase arrest by cytostatic factor: from establishment to release. Oncogene, 26(9): 1286-1289.
- Ludwig S., Engel K., Hoffmeyer A., Sithanandam G., Neufeld B., Palm D., Gaestel M., Rapp U.R. (1996). 3pK, a novel mitogen-activated protein (MAP) kinase-activated protein kinase, is targeted by three MAP kinase pathways. Molecular Cell Biology, 16(12): 6687-6697.
- Lolli G., Johnson L.N. (2005). CAK-Cyclin-dependent Activating Kinase: a key kinase in cell cycle control and a target for drugs? Cell Cycle, 4(4): 572-577.
- MacCorkle R.A., Tan T.H. (2005). Mitogen-activated protein kinases in cell-cycle control. Cell Biochemistry and Biophysics, 43(3): 451-461.
- Maher P. (1999). p38 mitogen-activated protein kinase activation is required for fibroblast growth factor-2-stimulated cell proliferation but not differentiation. The Journal of Biological Chemistry, 274(25): 17491-17498.
- Maller J.L., Schwab M.S., Gross S.D., Taieb F.E., Roberts B.T., Tunquist B.J. (2002). The mechanism of CSF arrest in vertebrate oocytes. Molecular and cellular Endocrinology, 187(1-2): 173-178.
- Manke I.A., Nguyen A., Lim D., Stewart M.Q., Elia A.E., Yaffe M.B. (2005). MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Molecular Cell, 17(1): 37-48.
- Marston A.L., Amon A. (2004). Meiosis: cell-cycle controls shuffle and deal. Nature Reviews. Molecular Cell Biology, 5(12): 983-997. Erratum in Nature Reviews. Molecular Cell Biology, 6(10): 818.
- McLaughlin M.M., Kumar S., McDonnell P.C., Van Horn S., Lee J.C., Livi G.P., Young P.R. (1996). Identification of mitogen-activated protein (MAP) kinase-activated protein kinase-3, a novel substrate of CSBP p38 MAP kinase. The Journal of Biological Chemistry, 271(14): 8488-8492.
- Meloche S., Pouysségur J. (2007). The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene, 26(22): 3227-3239.
- Mikalsen T., Gerits N., Moens U. (2006). Inhibitors of signal transduction protein kinases as targets for cancer therapy. Biotechnology Annual Review, 12: 153-223.
- Miller M.E., Cross F.R. (2001). Cyclin specificity: how many wheels do you need on a unicycle? Journal of Cell Science, 114(Pt 10): 1811-1820.
- Morgan D.O. (2007). In Primers in Biology: The Cell Cycle. Principles of Control. New Science Press Ltd and Oxford University Press.
- Mori M., Hara M., Tachibana K., Kishimoto T. (2006). p90Rsk is required for G1 phase arrest in unfertilized starfish eggs. Development, 133(9): 1823-1830.
- Myers A.P., Corson L.B., Rossant J., Baker J.C. (2004). Characterization of mouse Rsk4 as an inhibitor of fibroblast growth factor-RAS-extracellular signal-regulated kinase signaling. Molecular and Cellular Biology, 24(10): 4255-4266.
- Neufeld B., Grosse-Wilde A., Hoffmeyer A., Jordan B.W., Chen P., Dinev D., Ludwig S., Rapp U.R. (2000). Serine/Threonine kinases 3pK and MAPK-activated protein kinase 2 interact with the basic helix-loop-helix transcription factor E47 and repress its transcriptional activity. The Journal of Biological Chemistry, 275(27): 20239-20242.
- New L., Jiang Y., Zhao M., Liu K., Zhu W., Flood L.J., Kato Y., Parry G.C., Han J. (1998). PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO Journal, 17(12): 3372-3384.
- Ni H., Wang X.S., Diener K., Yao Z. (1998). MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochemical and Biophysical Research Communications, 243(2): 492-496.
- Nishiyama T., Ohsumi K., Kishimoto T. (2007). Phosphorylation of Erp1 by p90rsk is required for cytostatic factor arrest in Xenopus laevis eggs. Nature, 446(7139): 1096-1099.
- Paronetto M.P., Giorda E., Carsetti R., Rossi P., Geremia R., Sette C. (2004). Functional interaction between p90Rsk2 and Emi1 contributes to the metaphase arrest of mouse oocytes. EMBO Journal, 23(23): 4649-4659.
- Pawlowski W.P., Cande W.Z. (2005). Coordinating the events of the meiotic prophase. Trends in Cell Biology, 15(12): 674-681.
- Pei X.H., Xiong Y. (2005). Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene, 24(17): 2787-2795.
- Raman M., Chen W., Cobb M.H. (2007). Differential regulation and properties of MAPKs. Oncogene, 26(22): 3100-3112.
- Rausch O., Marshall C.J. (1999). Cooperation of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways during granulocyte colony-stimulating factor-induced hemopoietic cell proliferation. The Journal of Biological Chemistry, 274(7): 4096-4105.
- Reinhardt H.C., Aslanian A.S., Lees J.A., Yaffe M.B. (2007). p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11 (2): 175-189.
- Roux P.P., Blenis J. (2004). ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiology and Molecular Biology Reviews, 68(2): 320-344.
- Rowlett R.M., Chrestensen C.A., Nyce M., Harp M.G., Pelo J.W., Cominelli F., Ernst P.B., Pizarro T.T., Sturgill T.W., Worthington M.T. (2008). MNK kinases regulate multiple TLR pathways and innate proinflammatory cytokines in macrophages. Amercian Journal of Physiology - Gastrointestinal and Liver Physiology, 294 (2): G452-459.
- Rudolph J. (2007). Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry, 46(12): 3595-3604.
- Sabapathy K., Wagner E.F. (2004). JNK2: a negative regulator of cellular proliferation. Cell Cycle, 3(12): 1520-1523.
- Sapkota G.P., Kieloch A., Lizcano J.M., Lain S., Arthur J.S.C., Williams M.R., Morrice N., Deak M., Alessi D.R. (2001). Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90RSK and cAMP-dependent protein kinase, but not its farnesylation at Cys433, is essential for LKB1 to suppress cell growth. The Journal of Biological Chemistry, 276(22): 19469-19482.
- Schumacher S., Laass K., Kant S., Shi Y., Visel A., Gruber A.D., Kotlyarov A., Gaestel, M. (2004). Scaffolding by ERK3 regulates MK5 in development. EMBO Journal, 23(24): 4770-4779.
- Schwab M.S., Roberts B.T., Gross S.D., Tunquist B.J., Taieb F.E., Lewellyn A.L., Maller J.L. (2001). Bub1 is activated by the protein kinase p90(Rsk) during Xenopus oocyte maturation. Current Biology, 11(3): 141-150.
- Seternes O.M., Mikalsen T. Johansen B., Michaelsen E., Armstrong C.G., Morrice N.A., Turgeon B., Meloche S., Moens U., Keyse S.M. (2004). Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO Journal, 23(24): 4780-4791.
- Sha S.K., Sato T., Kobayashi H., Ishigaki M., Yamamoto S., Sato H., Takada A., Nakajyo S., Mochizuki Y., Friedman J.M., Cheng F.C., Okura T., Kimura R., Kufe D.W., VonHoff D.D., Kawabe T. (2007). Cell cycle phenotype-based optimization of G2-abrogating peptides yields CBP501 with a unique mechanism of action at the G2-checkpoint. Molecular Cancer Therapy, 6(1): 147-153.
- She Q.B., Ma W.Y., Dong Z. (2002). Role of MAP kinases in UVB-induced phosphorylation of p53 at serine 20. Oncogene, 21(10): 1580-1589.
- Sherr C.J. (2006). Divorcing ARF and p53: an unsettled case. Nature Reviews. Cancer, 6(9): 663-673.
- Sherr C.J., Roberts J.M. (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & Development, 13(12): 1501-1512.
- Silverman E., Frödin M., Gammeltoft S., Maller J.L. (2004). Activation of p90Rsk1 is sufficient for differentiation of PC12 cells. Molecular and Cellular Biology, 24(24): 10573-10583.
- Sinnett-Smith J., Zhukova E., Rey O., Rozengurt E. (2007). Protein kinase D2 potentiates MEK/ERK/RSK signaling, c-Fos accumulation and DNA synthesis induced by bombesin in Swiss 3T3 cells. Journal of Cellular Physiology, 211 (3): 781-790.
- Sithanandam G., Latif F., Duh F.M., Bernal R., Smola U., Li H., Kuzmin I., Wixler V., Geil L., Shrestha S. (1996). 3pK, a new mitogen-activated protein kinase-activated protein kinase located in the small cell lung cancer tumor suppressor gene region. Molecular and Cellular Biology, 16(3): 868-876.
- Smith J.A., Poteet-Smith C.E., Xu Y., Errington T.M., Hecht S.M., Lannigan D.A. (2005). Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Research, 65(3): 1027-1034.
- Sun M., Wei Y., Yao L., Xie J., Chen X., Wang H., Jiang J., Gu J. (2006). Identification of extracellular signal-regulated kinase 3 as a new interaction partner of cyclin D3. Biochemical and Biophysical Research Communications, 340(1): 209-214.
- Sun P., Yoshizuka N., New L., Moser B.A., Li Y., Liao R., Xie C., Chen J., Deng Q., Yamout M., Dong M.Q., Frangou C.G, Yates JR 3rd, Wright P.E., Han J. (2007). PRAK is essential for ras-induced senescence and tumor suppression. Cell, 128(2): 295-308.
- Taieb F.E., Gross S.D., Lewellyn A.L., Maller J.L. (2001). Activation of the anaphase-promoting complex and degradation of cyclin B is not required for progression from Meiosis I to II in Xenopus oocytes. Current Biology, 11(7): 508-513.
- Topisirovic I., Capili A.D., Borden K.L. (2002). Gamma interferon and cadmium treatments modulate eukaryotic initiation factor 4E-dependent mRNA transport of cyclin D1 in a PML-dependent manner. Molecular and Cellular Biology, 22(17): 6183-6198.
- Tunquist B.J., Schwab M.S., Chen L.G., Maller J.L. (2002). The spindle checkpoint kinase bub1 and cyclin e/cdk2 both contribute to the establishment of meiotic metaphase arrest by cytostatic factor. Current Biology, 12(12): 1027-1033.
- Tunquist B.J., Maller J.L. (2003). Under arrest: cytostatic factor (CSF)-mediated metaphase arrest in vertebrate eggs. Genes & Development, 17(6): 683-710.
- Ueda T., Watanabe-Fukunaga R., Fukuyama H., Nagata S., Fukunaga R. (2004). Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Molecular and Cellular Biology, 24(15): 6539-6549.
- van den Heuvel S. (2005). Cell-cycle regulation. WormBooks, September 21, 1-16.
- Vogelstein B., Lane D., Levine A.J. (2000). Surfing the p53 network. Nature, 408 (6810): 307-310.
- Voncken J.W., Niessen H., Neufeld B., Rennefahrt U., Dahlmans V., Kubben N., Holzer B., Ludwig S., Rapp U.R. (2005). MAPKAP kinase 3pK phosphorylates and regulates chromatin association of the polycomb group protein Bmi1. The Journal of Biological Chemistry, 280(7): 5178-5187.
- Vousden K.H. (2006). Outcomes of p53 activation--spoilt for choice. Journal of Cell Science, 119(Pt 24): 5015-5020.
- Weber H.O., Ludwig R.L., Morrison D., Kotlyarov A., Gaestel M., Vousden K.H. (2005). HDM2 phosphorylation by MAPKAP kinase 2. Oncogene, 24(12): 1965-1972.
- Wendel H.G., Silva R.L., Malina A., Mills J.R. Zhu H., Ueda T., Watanabe-Fukunaga R., Fukunaga R., Teruya-Feldstein J., Pelletier J., Lowe S.W. (2007). Dissecting eIF4E action in tumorigenesis. Genes & Development, 21(24): 3232-3237.
- Wenston C.R., Davis R.J. (2007). The JNK signal transduction pathway. Current Opinion in Cell Biology, 19(2): 142-149.
- Wong K., Zhang J., Awasthi S., Sharma A., Rogers L., Matlock E.F., Van Lint C., Karpova T., McNally J., Harrod R. (2004). Nerve growth factor receptor signaling induces histone acetyltransferase domain-dependent nuclear translocation of p300/CREB-binding protein-associated factor and hGCN5 acetyltransferases. The Journal of Biological Chemistry, 279(53): 55667-55674.
- Worch J., Tickenbrock L., Schwäble J., Steffen B., Cauvet T., Mlody B., Buerger H., Koeffler H.P., Berdel W.E., Serve H., Müller-Tidow C. (2004). The serine-threonine kinase MNK1 is post-translationally stabilized by PML-RARalpha and regulates differentiation of hematopoietic cells. Oncogene, 23(57): 9162-9172.
- Wu J.Q., Hansen D.V., Guo Y., Wang M.Z., Tang W., Freel C.D., Tung J.J., Jackson P.K., Kornbluth S. (2007). Control of Emi2 activity and stability through Mos-mediated recruitment of PP2A. The Proceedings of the National Academy of Sciences of the United States of America, 104(42): 16564-16569.
- Xiao Z., Chen Z., Gunasekera A.H., Sowin T.J., Rosenberg S.H., Fesik S., Zhang H. (2003). Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. The Journal of Biological Chemistry, 278(24): 21767-21773.
- Xiao Z., Xue J., Sowin T.J., Zhang H. (2006). Differential roles of checkpoint kinase 1, checkpoint kinase 2, and mitogen-activated protein kinase-activated protein kinase 2 in mediating DNA damage-induced cell cycle arrest: implications for cancer therapy. Molecular Cancer Therapeutics, 5(8): 1935-1943.
- Yannoni Y.M., Gaestel M., Lin L.L. (2004). P66(ShcA) interacts with MAPKAP kinase 2 and regulates its activity. FEBS Letters, 564(1-2): 205-211.
- Yawsen P., Campisi J. (2007). Oncogene-induced senescence pathways weave an intricate tapestry. Cell, 128(2): 233-234.
- Zaru R., Ronkina N., Gaestel M., Arthur J.S., Watts C. (2007). The MAPK-activated kinase Rsk controls an acute Toll-like receptor signaling response in dendritic cells and is activated through two distinct pathways. Nature Immunology, 8(11): 1227-1235. Corrigendum in Nature Immunology (2008), 9(1): 105.
- Zhang W., Liu H.T. (2002). MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Research, 12(1): 9-18.
- Zhang Y., Dong C. (2007). Regulatory mechanisms of mitogen-activated kinase signaling. Cellular and Molecular Life Sciences, 64(21): 2771-2789.