





Publications
Presentations/Lectures
Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility
Nancy Gerits, Sergiy Kostenko, Alexey Shiryaev, Mona Johannessen, Ugo Moens*
Faculty of Medicine, Department of Microbiology and Virology, University of Tromsø, N-9037 Tromsø, Norway
*Corresponding author: Ugo Moens, phone: +47-77644622, fax: +47-77645350, e-mail: ugom@fagmed.uit.no
Keywords: cross-talk; PKA; MAPK; ERK; p38; JNK; compartmentalized signalling; PDE
Abstract:
Inter- and intracellular communications and responses to environmental changes are pivotal for the orchestrated and harmonious operation of multi-cellular organisms. These well-tuned functions in living organisms are mediated by the action of signal transduction pathways, which are responsible for receiving a signal, transmitting and amplifying it, and to elicit the appropriate cellular responses. Mammalian cells posses numerous signal transduction pathways that, rather than acting in solitude, interconnect with each other, a phenomenon referred to as cross-talk. This allows cells to regulate the distribution, duration, intensity and specificity of the response. The cAMP/cAMP-dependent protein kinase (PKA) pathway and the mitogen-activated protein kinase (MAPK) cascades modulate common processes in the cell and multiple levels of cross-talk between these signalling pathways have been described. The first and best-characterized interconnections are the PKA-dependent inhibition of the MAPKs ERK1/2 mediated by RAF-1, and PKA-induced activation of ERK1/2 interceded through B-RAF. Recently, novel interactions between components of these pathways and new mechanisms for cross-talk have been elucidated. This review discusses both known and novel interactions between compounds of the cAMP/PKA and MAPKs signalling pathways in mammalian cells.
1 Introduction
An essential way that guaranties the harmony and normal functioning of cells in a multi-cellular organism lies in the ability of cells to properly communicate with each other. Cellular communication occurs by exchanging signals (molecules or ions) that bind specifically to surface or intracellular receptors. Binding of a signal to its cognate receptor triggers a cascade of events that amplifies and transmits the incoming signal, and eventually elicits a cellular response. This signal transduction often requires the orchestrated action of protein kinases, which by sequential phosphorylation events enable conduction and finally translation of the signal into a cellular reaction [1,2]. Two of the well-characterized signal transduction pathways are the cAMP/cAMP-dependent protein kinase (PKA) and the mitogen-activated protein kinase (MAPK) pathways. As with many other signalling pathways, the PKA and MAPK pathways do not operate independently of each other, rather they interrelate with each other, commonly referred to as cross-talking [3-5]. Cells are constantly exposed to numerous stimuli from their environment and cross-talk between signalling pathways allows the cell to distribute the incoming signals and fine-tune the responses. The interaction between signal transduction pathways can be partially compared with a football game where the players in a team will form a relay to transmit the signal (pass the ball) to obtain their goal (to score). At the same time, players from the other team will try to neutralize the opponents and intercept the ball. However, a major difference between a football contest and signal transduction is that the compounds in different signalling pathways (opponents in the different teams) actually can collaborate to obtain a common goal. The cross-talk between PKA and the MAPK kinase pathway has been the topic of two previous excellent reviews [3,5]. This review updates and expands the earlier reports on the different interactions between the PKA and MAPK signalling pathways in mammalian cells. We will briefly introduce the "PKA team" with its more than 50 distinct "players" including e.g. the subunits that constitute PKA, cAMP generating and degenerating enzymes (adenylyl cyclases and phosphodiesterases), PKA anchoring proteins, and the "MAPK team" with its great diversity of players. We will then focus on the different strategies and levels of cross-talk between these pathways and discuss the outcome of the interconnections between these pathways.
2 The cAMP/PKA and MAPK pathway
2.1 The cAMP pathway
Extracellular signals are often converted into an intracellular signal referred to as a second messenger. Adenosine 3',5' cyclic monophosphate (cAMP) was one of the first secondary messengers identified in cells. Adenylyl cyclases, a large family of proteins that are regulated by trimeric G-proteins linked to G-protein coupled receptors, use ATP to generate cAMP. The initial molecule purified to bind cAMP and probably the major target of cAMP was protein kinase A (PKA) or cAMP-dependent protein kinase [reviewed in 6 and 7]. PKA is a serine/threonine kinase that in its inactive form consists as a tetramer of two regulatory subunits (R) and two catalytic subunits (C). Each R subunit contains two binding sites for cAMP. Upon binding of cAMP to the R subunits, the PKA holoenzyme dissociates and the two C subunits, which possess the protein kinase activity, are released (see Figure 1). The mammalian genome encodes three genes for the catalytic isoforms (Cα, Cβ, and Cγ), and four genes for the regulatory subunits (RIα, RIβ, RIIα, and RIIβ), allowing the formation of different compositions of PKA holoenzymes. The RI-containing holoenzymes are predominantly cytoplasmic, while RII-containing PKAs preferentially associate with scaffold proteins known as the A-kinase anchoring proteins or AKAPs [8-12].
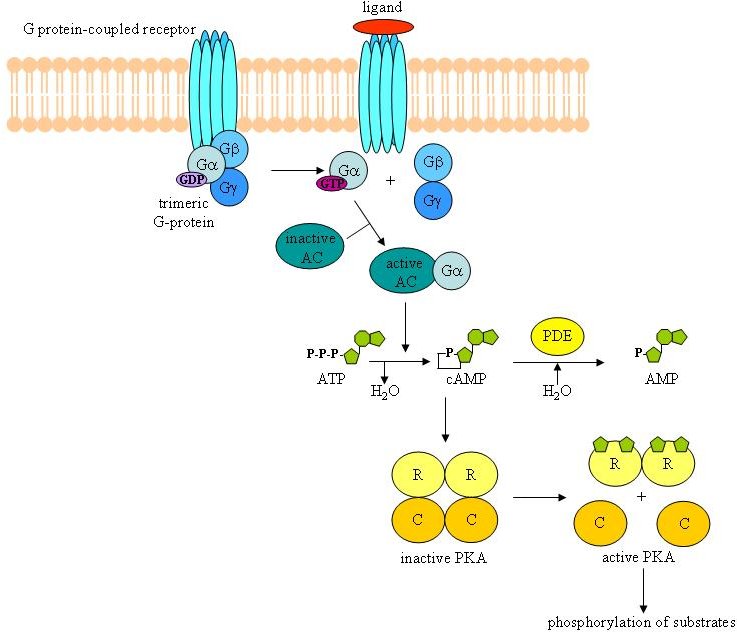
Figure 1. Schematic diagram of the cAMP/PKA signalling pathway. Upon activation of a G-protein coupled receptor, the trimeric Gαβγ protein dissociates into the active Gα subunit, loaded with GTP. The GTP-loaded Gα will activate adenylyl cyclase (AC), which generates cAMP from ATP. Next, cAMP binds to the regulatory subunits (R) of the PKA and induces dissociation of the holoenzyme. The catalytic subunits (C) can then phosphorylate their substrates. Phosphodiesterases (PDE) hydrolyse cAMP into AMP. This represents one way to shut down the activated cAMP/PKA pathway.
Other than PKA, phosphodiesterases (PDE) can also bind cAMP. These enzymes control the levels of cAMP by catalyzing the conversion of cAMP into AMP [5,6 Lugnier, 2006]. This degradation of cAMP comprises one of the feedback mechanisms that can shut down the cAMP/PKA signalling pathway. In addition, PDE serve as targets for crosstalk by the MAPK ERK1/2. Because of the central role of PDE in different PKA-MAPK interactions, their structure, biological role and cross-talk will be more extensively discussed in a separate section (section 3.1.9.).
Other cAMP effectors include some cyclic nucleotide-gated channels and the Epac (exchange proteins directly activated by cAMP) 1 and 2 proteins, also known as cAMP-regulated guanine nucleotide exchange factors 1 and 2 (cAMP-GEFI and cAMP-GEFII). Epac1 and Epac2 were characterized in 1998 by two independent research teams [reviewed in 16]. These guanine exchange factors facilitate the GDP/GTP exchange for the small GTPase proteins Rap1 and Rap2. Rap1 interacts with the serine/threonine kinases RAF1 and B-RAF, establishing a link between cAMP and MAPK signalling (see section 3.1.6.). Another guanine-exchange factor (GEF), which possesses cAMP/cGMP-binding domains and can be activated by cAMP is CNrasGEF [17,18].
Finally, additional database searches for proteins with cAMP-binding domains have identified novel putative effectors, but their role in cAMP signalling has been poorly investigated and hence their potential connections with the MAPK cascades remain elusive [16,19,20].
2.2 The mitogen-activated protein kinase (MAPK) pathways
As the name suggests, mitogens were the first group of signals identified that stimulated these pathways [21]. Other signals that elicit MAPK activation include stress and cytokines [for a recent review see 22]. MAPK pathways regulate cellular processes such as proliferation, survival/apoptosis, differentiation, development, adherence, motility, metabolism, and gene regulation. The classical mitogen-activated protein kinase (MAPK) signalling pathways consist of cascade of three consecutive phosphorylation events exerted by a MAPK kinase kinase (MAPKKK), a MAPK kinase (MAPKK), and a MAPK. MAPK in turn phosphorylates non-protein kinase substrates or yet other protein kinases. The latter are referred to as MAPK-activated protein kinases (MAPKAPK). Figure 2 illustrates the different MAPK signalling modules identified in mammalian cells, with the three major ones RAF>MEK>ERK, the JNK, and the p38 MAPK pathways [22-29].
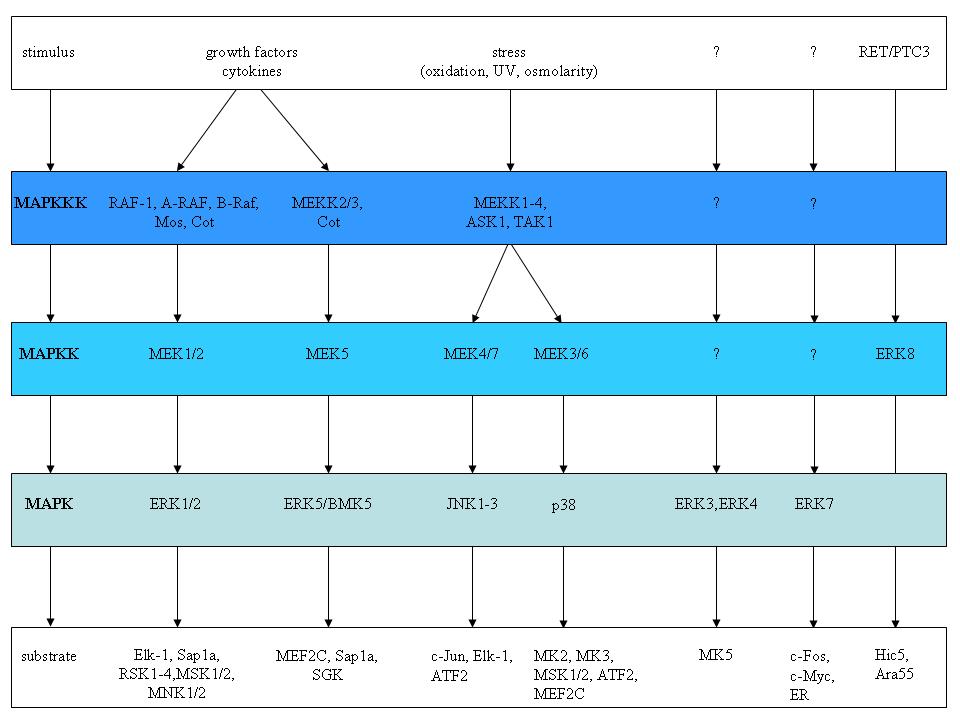
Figure 2. The different MAPK cascades in mammalian cells. The typical MAPK pathway consists of a tripartite module in which a MAPKKK phosphorylates a MAPKK, which in turn phosphorylates a MAPK. The nature of the signals that activate the different MAPK cascades are shown at the top of the figure, while some of the substrates are indicated at the bottom of the figure. The poorly identified atypical ERK3, ERK4, ERK7 and ERK8 pathways are also depicted. The figure is adapted from [16].
In general, MAPK pathways are not directly coupled to receptors. In stead they require effector molecules that transmit the signal of the activated receptor to specific pathways. These effectors serve as popular targets for cross-talk [30]. One important family of effectors of the RAF>MEK>ERK pathway that plays a key role in activation of this pathway and that is also involved in some of the cross-talk with the cAMP-signalling pathway is briefly discussed here. These effectors are a family of small guanine nucleotide-binding proteins (G-proteins) that are activated when loaded with GTP and inactive in a GDP-associated state. The prototype of this family is the p21 RAS protein, referred hereafter as RAS. The GTP/GDP occupancy of RAS is catalyzed by GEFs. Both RAS and GEF activity is, as outlined in 3.1.2., regulated by cAMP/PKA-dependent and cAMP/PKA-independent cascades [31].
Many proteins in the MAPK cascades exist as different isoforms due to alternative splicing, or as highly sequence-related proteins. Despite their high sequence homology, these proteins regularly exert non-overlapping functions and studies with knock-out mice have shown that they are often non-redundant [reviewed in 32 and 33]. E.g. the RAF MAPKKK family consists of the members RAF-1, A-RAF, and B-RAF. RAF-1 is ubiquitously expressed, but A-RAF and B-RAF are differentially expressed with most abundant levels in urogenital and in neural tissues, respectively. In addition, B-RAF exists in different isoforms, 68 kDa and 95 kDa being the two major variants [34]. While general knock-out of one of the raf-genes (A-RAF, B-RAF, or RAF-1 deficient animals) always resulted in lethality, tissue-specific disruption of the different raf genes generated mice with dissimilar phenotypes. This indicates that the RAF isoforms cannot functionally compensate for each other. Discussion of the structure, functions, and knock-out phenotypes of each MAPK in the MAPK cascades is beyond the scope of this survey, and the reader is referred to recent reviews on these topics [23-29,32].
3 cAMP/PKA-MAPK interactions
The PKA and MAPK pathways do not operate independently of each other, but multiple cross-talk events between these pathways can occur. The known interactions between these two pathways are summarized in Figures 3A and 3B, and will be discussed in detail in this section. The reader should be aware that some interactions are cell-specific and the figure merely idealizes all the possible interactions that have been described, rather than depicting the true in vivo situation. Moreover, the PKA and MAPK pathways do not always interact as exemplified, in immature (one day-old) primary neuronal cultures from rat [35]. At the time point where the initiation of neurite formation is maximal, no interaction was observed, while in more mature (7 days old) cultures, where synaptic contacts have been established, a weak but reproducible activation of MAPK following stimulation of the cAMP/PKA pathway was monitored. These results suggest that cAMP/PKA and MAPK signalling act independently at the initiation of neuritogenesis but become coupled during later stages of neuronal development [35]. To add to the complexity, cAMP/PKA-MAPK cross-talk can also be restricted to specific intracellular microenvironments, allowing compartmentalized signalling (see further). Although our understanding of the mechanisms of cross-talk has increased enormously, some points of dispute remain and not all molecular mechanisms have been solved. In the following part we will review the various mechanisms by which the cAMP/PKA and MAPK "teams" collaborate or impede each other, thus illustrating their "comradeship and hostility".
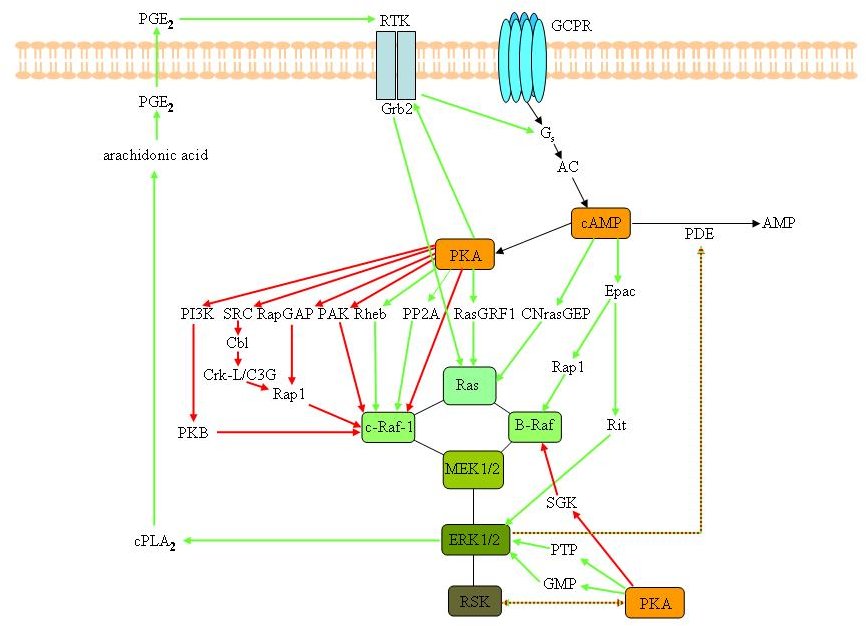
Figure 3A. Possible cross-talk events between the cAMP/PKA and the MAPKs signalling cascades. Interactions between the RAS/RAF/MEK/ERK and cAMP/PKA signalling pathways in mammalian cells. cAMP generated by adenylyl cyclases (AC) can affect the MAPK pathways in a PKA-dependent and -independent manner. Depending on the cell type, cAMP/PKA can decrease or increase the activity of MAPKs.
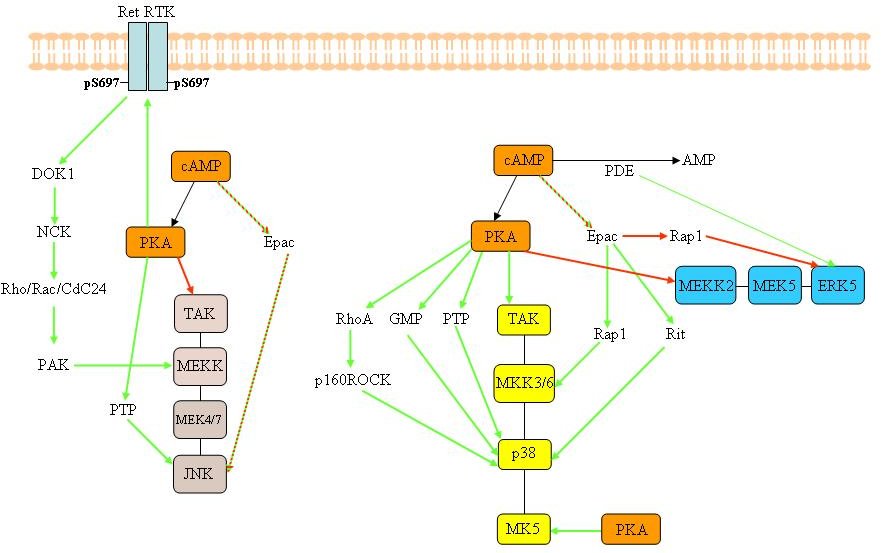
Figure 3B. Possible cross-talk events between the cAMP/PKA and the MAPKs signalling cascades. Cross-talk between other MAPKs signalling cascades and cAMP/PKA. The green arrows indicate that the interaction leads to activation of the target, while red arrows illustrate negative interactions. A two-coloured arrow indicates that the interaction can stimulate or inhibit the target protein. We refrained from showing the common downstream substrates between PKA and components of the MAPK pathways in this figure. GPCR= G-protein coupled receptor; PDE= phosphodiesterase; RTK= receptor tyrosine kinase. The reader is refereed to the text for more details.
3.1 Mechanisms of cAMP/PKA and MEK/ERK cross-talk
3.1.1 Introduction
In 1993, five independent groups almost simultaneously reported for the first time interconnection between the RAS-RAF-MEK-ERK and the cAMP-PKA pathways. They all demonstrated that cAMP blocked growth signal-induced activation of the MEK/ERK signalling cascade in fibroblasts, fat cells, and smooth muscle cells. They also pinned down the location of this inhibition somewhere between RAS and c-RAF, but the exact mechanism remained unsolved at that time [36-40]. However, at the same time, other studies revealed that elevated cAMP levels stimulated MEK/ERK activity in PC12 cells [41,42], while other researcher observed no effect of cAMP on ERK1/2 activity in primary cultures of dog thyroid epithelial cells and in primary sympathetic neurons [43,44]. In one study in CCL39 cells, cAMP delayed, rather than inhibited, ERK1/2 activation [45]. To add to the complexity, Erhardt and co-workers found that in PC12 cells maintained in serum-containing medium, B-RAF was refractory to inhibition by cAMP, whereas RAF-1 was effectively inhibited, indicating reverse effects of cAMP on ERK1/2 activity [46]. Soon after the discovery of the PKA-ERK1/2 interactions, it was established that the opposite effects of cAMP/PKA on ERK1/2 activation in different cell lines could be explained by the dissimilar involvement of the RAF isoforms RAF-1 and B-RAF or splice variants [4,47 and references therein]. Originally, it was assumed that cAMP/PKA-triggered inhibition of ERK1/2 in a cell-type seemed to be mediated by RAF-1, while induction of ERK1/2 by cAMP/PKA in another cell-type involved B-RAF. The use of dominant negative variants of RAF isoforms, RNA interference, or cells deficient in specific RAF isoforms may help to determine the putative involvement of a particular RAF protein in the PKA-ERK1/2 interaction. Alternatively, assays to measure the activity of the different RAF isoforms in response to activation of the PKA pathway may also shed light on the possible contribution of the individual RAF proteins. Yet another approach could be monitoring the subcellular localization of the RAF isoforms or perform FRET analysis to study the cAMP-induced interaction between RAF isoforms and target proteins such as RAS or MEK1/2 in cAMP-treated cells. Apparently, multiple mechanisms by which RAF isoforms mediate cross-talk between PKA and ERK seem to be operational and these will be outlined here.
3.1.2 cAMP/PKA-RAS interconnection
RAS is the prototype of the small GTPase proteins, a family of proteins whose activity is stimulated by replacing GDP with GTP [48]. In its GTP-loaded/activated state, RAS serves as a master regulator of several signalling pathways, including the MEK1/2-ERK1/2 pathway through RAF [49,50]. Despite the original studies, which indicated that RAS is not involved in the activation of ERK1/2 by PKA, some groups have later shown a role for RAS in cAMP-induced activation of ERK1/2 in AtT20 cells, selected cultured cortical neurons, hippocampal neurons and in melanocytes. By use of PKA inhibitors, PKA-independent and -dependent manners of cAMP-provoked RAS activation have been described [17,51-55]. One mechanism by which cAMP can modulate RAS was suggested by Pham and colleagues [17,18]. They demonstrated that the RAS guanine nucleotide exchange factor CNrasGEF is involved in cAMP-induced RAS activation. This GEF possesses cAMP/cGMP-binding domains, and can be activated in a cAMP-dependent, but PKA-independent manner. Another GEF that may be engaged in the cAMP/PKA-Ras interaction is RasGRF1. This GEF could be directly phosphorylation by PKA on Ser-927 [56], but the responsiveness of GEF to cyclic nucleotides has been challenged [57]. Moreover, elevated cAMP has been shown to block RAS-binding to B-RAF, but the effect on ERK1/2 was not investigated [58].
3.1.3 Phosphorylation of RAF-1 by PKA
RAF-1 activation necessitates membrane recruitment and functional interactions with many proteins, including kinases (SRC, PAK, PKA, PKB, PKC, and B-RAF), phosphatases (PP1, PP2A), and scaffolding proteins (14-3-3, Hsp90, KSR, and RKIP) [59,60]. In addition, RAF-1's activity is controlled by complex phosphorylation events, which will be briefly described here. For more extensive information, the reader is referred to [60-63]. In resting cells, RAF-1 is constitutively phosphorylated at Ser-259 and Ser-621. This allows 14-3-3 proteins to bind these sites and to trap RAF-1 in an inactive, closed conformation [61,64]. In addition, the A subunit and the catalytic C subunit of the trimeric protein phosphatase 2A (PP2A) are in complex with RAF-1. Stimuli like growth factors activate RAS, and induce recruitment of the B subunit of PP2A. The B subunit triggers activation of PP2A, and subsequent dephosphorylation of RAF-1 at phosphoSer-259 [65]. Dephosphorylation of this site displaces 14-3-3 and leads to an open conformation and recruitment of RAF-1 to the plasma membrane for full activation. RAF-1 can then phosphorylate MEK1/2, which results in phosphorylation and activation of ERK1/2 [66]. When cAMP levels increase, activated PKA will phosphorylate RAF-1 on residues Ser-43 and Ser-233 [66,67]. Phosphorylation of the latter residue creates a new 14-3-3 binding site that switches 14-3-3 from binding phosphoSer-621 to occupying phosphoSer-233. The reason for this switch may lie in a higher affinity of 14-3-3 for phosphoSer-233 than phosphoSer-621 because of easier accessibility due to a favourable conformation of the RAF-1 protein [61]. 14-3-3 switching locks RAF-1 in an inactive form. Moreover, phosphorylation of Ser-43 causes steric hindrance, which has been suggested to reduce the affinity of RAF-1 with RAS-GTP [61,68]. Both actions will prevent membrane recruitment of RAF-1 and hence activation of the RAF-MEK1/2-ERK1/2 cascade [3,59-61,63,64,68,69]. However, the exact mechanism of RAF-1 inhibition by PKA remains a point of dispute. E.g. in contrast to the above findings, another study in HEK293 cells demonstrated that substitution of Ser-43 into the non-phosphorylable Ala or deletion of the N-terminal 50 amino acids of RAF-1 did not impede cAMP-dependent inhibition of epidermal growth factor-induced RAF-1 kinase activity when this mutant was overexpressed. This result indicates that Ser-43 phosphorylation by PKA is not necessary for the inhibition of RAF-1 activity by PKA in vivo [67]. An alternative or additional mechanism for PKA-induced inhibition of ERK signalling pathway may involve RAF-1 phosphorylation at still another residue. Indeed, PKA blocked EGF-induced ERK1/2 activation through inhibition of RAF-1 phosphorylation at Ser-338. PAK can phosphorylate RAF-1 on Ser-338, while PKA can phosphorylate and inhibit PAK. Thus, one mechanism by which PKA can inhibit RAF-1 is by inhibiting PAK and thus preventing phosphorylation of RAF-1 at Ser-338. This model is underscored by the observation that RAF-1 Ser-338 phosphorylation provoked by ectopic expression of an activated PAK variant is counteracted by forskolin [70]. Finally, PKA-mediated RAF-1 phosphorylation at Ser-621 has also been suggested as a mechanism by which PKA can interfere with ERK1/2 signalling [71]. PKA was shown to phosphorylate RAF-1 at Ser-621 and this strongly reduced RAF-1 kinase activity in vitro, suggesting that PKA could negatively interfere with ERK1/2 activation by hampering the enzymatic activity of RAF-1. Furthermore, co-expression of the catalytic subunit of PKA and wild-type RAF-1 ablated epidermal growth factor (EGF)-induced RAF-1 activity, while mutations in Ser-621 or substitution of Arg-618 into fenylalanine, which destroys the PKA recognition site, rendered these RAF-1 mutants refractory to PKA inhibition [71]. However, phosphorylation of Ser-621 in vitro is ~100-fold less efficient than Ser-43, and evidence for in vivo phosphorylation of Ser-621 by PKA is lacking. These observations and the knowledge that Ser-621 is constitutively phosphorylated jeopardize the role of this residue as a bona fide PKA phosphorylation site, and keep the functional implication of Ser-621 in PKA-induced inhibition of RAF-1 controversial [67].
PKA may also affect RAF-1 activity indirectly by phosphorylating RAF-1-interacting proteins. The protein phosphatase 2A (PP2A) is activated by PKA through phosphorylation of Ser-566 in the B56δ subunit [72]. Upon dephosphorylation of Ser-259 by PP2A, RAF-1 is recruited to the membrane and binds to RAS. Thus PKA may, through activation of PP2A, activate RAF-1, but biological evidence is lacking.
3.1.4 Phosphorylation of B-RAF through the cAMP/PKA pathway
Despite their high sequence homology between the RAF-1 and B-RAF isoforms (81% identity within their catalytic domain), B-RAF is probably not a direct PKA substrate for the interference of PKA with the MEK/ERK pathway [73]. PKA can phosphorylate B-RAF in vitro and in vivo, but the phosphoacceptor sites have not been accurately mapped. B-RAF lacks the equivalent of Ser-43 in RAF-1, while Ser-728 (in the 95 kDa isoform) corresponds to Ser-621 in RAF-1. Ser-728 may be target for PKA as its flanking sequences are highly homologous to those in RAF-1, but this remains to be established [61]. The consequences of PKA-mediated B-RAF phosphorylation have been poorly investigated, but all three 14-3-3 binding sites of RAF-1 are conserved in B-RAF. Ser-728 is a 14-3-3 binding site that appears to be necessary for the biological activity of B-RAF, while binding of 14-3-3 to Ser-365 seems to be essential to keep B-RAF in an inactive conformation [74]. The study by Qiu and co-workers suggested that the amount of 14-3-3 bound to B-RAF could influence the outcome of PKA on B-RAF activity [47]. The authors explored the role of 95 kDa B-RAF and 14-3-3 in cAMP-induced modulation of the MEK/ERK in four different cell lines: PC12, CHO-K1, C6, and NB2A. As previous mentioned (section 2.2), 68 kDa and 95 kDA are the two major isoforms of B-RAF. All four cells lines expressed comparable levels the 95 kDa splice variant of B-RAF, and the levels of the 68 kDa variant were similar to those of 95 kDa B-RAF in C6 and NB2A, while PC12 and K1 contained lower but comparable concentrations of 68 kDa than the 95 kDa isoform. Moreover, these four cells possessed similar amounts of 14-3-3 proteins. The authors focused only on the role of the 95 kDa B-RAF isoform. While cAMP activated Rap1 in all tested cell lines, B-RAF and ERK1/2 were stimulated in PC12 and CHO.K1 cells, but inhibited in C6 and NB2A cells. Similar results were obtained with cells maintained in medium with low or high serum concentrations. These results indicate that Rap1 activation is not sufficient to activate B-RAF in C6 and NB2A cells. The authors found that 5-fold less 14-3-3 was associated with B-RAF in cells in which cAMP was inhibitory compared to cells in which cAMP was stimulatory, despite comparable 14-3-3 amounts in all four cell lines. Overexpression of 14-3-3 prevented B-RAF inactivation by cAMP, while ectopic expression of the non-B-RAF binding 14-3-3 mutant resulted in partial loss of cAMP-induced B-RAF activation. These results suggest that 14-3-3 occupancy of B-RAF protects this kinase from inactivation by PKA [47 and references therein]. The effect of PKA-induced B-RAF phosphorylation on 14-3-3 binding switch has not been studied in detail, but a similar regulatory mechanism as in RAF-1 may be operational.
Another mechanism by which PKA may regulate the B-RAF-ERK1/2 cascade is the assumption that PKA may affect the kinase activity of B-RAF. In co-transfection studies in PC12 cells, PKA activated full-length B-RAF, but inhibited the isolated catalytic domain, while in vitro PKA had no effect on full-length B-RAF activity, but reduced the activity of the catalytic domain [75]. Phosphorylation of Thr598 and Ser601 is essential for B-RAF activation by RAS, but whether they are target for PKA has not been established [76]. It is also possible that PKA phosphorylates other proteins which may alter B-RAF's activity, but this remains to be confirmed.
3.1.5 Rap-1-mediated inhibition of the RAF-1-MEK1/2-ERK1/2 signalling pathway by PKA
Another means by which PKA may prevent RAS-dependent RAF-1 activation involves the small GTPase Rap1. Rap1 was identified as an inhibitor of ERK1/2 in Rat-1 fibroblasts [77]. The actions of Rap1 manifest themselves upstream of RAF-1, but downstream of RAS because Rap1 could block constitutively activated RAS, but not constitutively activated RAF-1 [78]. It was shown that activation of endogenous Rap1 blocked RAS binding to RAF-1 and that PKA could directly phosphorylate Rap1 at Ser-179, which increased GTP-loading. Moreover, the Rap1 inactivator RapGAP can also be phosphorylated in vivo by PKA [79-82]. However, the consequences of direct PKA phosphorylation on Rap1's activity remained elusive as this modification is not required for the activation of Rap1 by cAMP [78,82,83].
Recently, the mechanism by which PKA governs Rap1 activity to inhibit the ERK1/2 signalling pathway was resolved, and will be outlined here. As a start, it should be mentioned that Klinger and colleagues originally had found that cAMP generated after stimulation of the A2A-adenosine receptor resulted in activation of PKA and stimulation of ERK1/2 in different cell lines, including CHO, PC12, and NIH3T3 cells endogenously or ectopically expressing this receptor [84]. SRC family kinases acting downstream of PKA were necessary for induction of ERK1/2 upon agonist binding to the A2A-adenosine receptor as blockage of SRC family kinases blunted ERK1/2 phosphorylation. Diminished ERK activation was also monitored in a forskolin-treated mouse embryonic fibroblast cell line devoid of the SRC kinases SRC, YES and FYN. Reintroduction of c-SRC, but not the S17A mutant which cannot be phosphorylated by PKA in these cells restored ERK1/2 activation upon forskolin exposure [84]. The missing pieces in the puzzle that links PKA, SRC, and ERK1/2 were later filled in by others to give following complete picture. Increased cAMP levels activate PKA, which directly phosphorylates the tyrosine kinase SRC at Ser-17 [85]. This triggers autophosphorylation of Tyr-418. This activation of SRC leads to tyrosine phosphorylation of the downstream effector Cbl and subsequent recruitment of the Crk-L/C3G (=Crk Src homology domain 3 guanine nucleotide exchange factor) complex via the SH2 domain of Crk-L [86]. The Rap1-GEF C3G contributes in GTP-loading of Rap1 [87]. The GTP-bound form of Rap1 can then bind to the N-terminal regulatory domain of RAF-1, sequester it from RAS, thereby blocking RAS/RAF-1/MEK/ERK signalling [75 and references therein].
3.1.6 Rap-1-mediated activation of the B-RAF-MEK1/2-ERK1/2 signalling pathway by cAMP/PKA
Our understanding of the mechanisms by which cAMP induced activation of ERK1/2 through B-RAF expanded by the identification of the small GTPase Rap1 and the Rap1-GEFs that can be directly activated by cAMP, Epac1 and Epac 2 (also known as cAMP-GEF-I and cAMP-GEF-II) [16]. It was generally accepted that cAMP bound directly to Epac, and that the cAMP-loaded Epac then served as a GTP exchange factor allowing GDP-GTP exchange and concomitant activation of Rap1. Active GTP-Rap1 in turn activated B-RAF, which led to a phosphorylation cascade of MEK1/2 and ERK1/2 [82,88]. Thus despite the high sequence homology between RAF-1 and B-RAF, Rap1 seems to exert opposite effects on these two protein kinases: inhibition of RAF-1, but stimulation of B-RAF. This model elegantly explains how cAMP hampered ERK1/2 via RAF-1, while it induced ERK1/2 through B-RAF.
Despite the refinement of the model, several observations questioned the involvement of cAMP-triggered Rap1 in B-RAF and ERK1/2 activation. Qiu and co-workers failed to monitor B-RAF activation by cAMP despite Rap1 activation [47]. By the use of the recently developed specific Epac inhibitor 8-(4-chloro-phenylthio)-2'-O-methyladenosine-3',5'-cyclic monophosphate (8CPT-2Me-cAMP), it was shown that both in cell lines in which cAMP inhibits growth-factor-induced ERK1/2 activation and in which cAMP activates ERK, Epac was incapable of activating ERK1/2 [89-91]. The dispute whether Rap1 activated by cAMP-Epac can induce ERK1/2 activation was recently solved by the work of the group of Stork [91]. In a series of studies, they showed how compartmentalized signalling enabled Epac/Rap1 to activate certain subcellular pools of ERK1/2. The incapability of Epac to activate ERK1/2 appeared to be a consequence of its subcellular localization. Upon cAMP stimulation, C3G, as well as Rap1, are localized to the plasma membrane, thus allowing activation of the physiological target of Rap1, B-RAF. Activated B-RAF can than activate the MEK1/2-ERK1/2 pathway. On the other hand, Epac resides perinuclear and is not recruited to the plasma membrane upon cAMP stimulation, hence the pools of Rap1 activated by Epac are not appropriately localized to couple to the B-RAF-MEK1/2-ERK1/2 signalling pathway. Thus PKA-dependent activation of Rap1 at the membrane occurs via C3G and leads to activation of ERK1/2, while PKA-independent activation of Rap1 in the perinuclear region proceeds via Epac and is incapable of activating ERK1/2 [91]. This is not the end of the story, because recently it was shown in lung carcinoma H1299 cells that EGF or cAMP elevating agents alone failed to promote significant translocation of Epac2 from the cytosol to the plasma membrane, while co-challenge with these stimuli induced redistribution of Epac2 to the plasma membrane and activation of a membrane-enriched pool of Rap1. This redirection of Epac2 to the plasma membrane was independent of PKA and was also observed by the specific Epac activator 8CPT-2Me-cAMP [92]. Although not tested by the authors, these findings suggest that ERK1/2 can be activated by cAMP through Epac2 and Rap1. Compartmentalized signalling can also explain PKA-dependent activation of ERK1/2 in rat cerebellar cells. In these cells, forskolin or pituitary adenylate cyclase activating protein (PACAP) activated ERK1/2. A dual pathway seemed to be involved. Increased cAMP levels led to activation of PKA, which could then activate ERK1/2 via RAS or through Rap1 in a SRC-dependent manner. The mechanism by which PKA activates RAS is unknown, but putative models have been described in section 3.1.2. Activated Rap1 regulated the activity of ERK1/2 in membrane fractions, but not in cytosolic fractions [93]. In agreement with the compartmentalized signalling suggested by the work of the laboratory of Stork, Morozov and colleagues reported that Rap1 also mediated ERK activation in membrane fractions of hippocampal neurons, but not in cytosolic fractions or total cell lysates [94].
To add to the complexity of Rap1's role in the cAMP/PKA-ERK1/2 cross-talk, Wang and colleagues observed that the mode of Rap1 activation by cAMP seems to be cell-specific [91]. They found that forskolin-induced Rap1 activation in the rat pheochromocytoma cell line PC12 was prevented by PKA inhibition, while PKA inhibition enhanced forskolin-triggered Rap1 activation in the mouse pituitary tumour cell line AtT20. Accordingly, they showed that C3G mediates all Rap1 activation by cAMP in PC12 cells, while Epac is the main target of cAMP-provoked activation of Rap1 in AtT20 cells. The observation that AtT20 cells express high levels of Epac1 and Epac2, while PC12 cells have low or undetectable levels of Epac1 and Epac2, respectively, underscores their findings [91].
There are also indications that cAMP can inhibit Rap1 [95]. Studies in rat C6 glioma cells revealed that cAMP inhibited ERK activity. This cAMP-hampered ERK activity occurred through inhibition of Rap1, but was PKA- and Epac-independent because the ERK1/2 phosphorylation levels were still reduced by cAMP in the presence of the PKA inhibitor H-89 or the constitutive active EpacΔcAMP (a mutant that lacks the cAMP-binding domain). In fact, EpacΔcAMP increased the phosphorylation levels of ERK1/2. The authors suggested two possible mechanisms by which cAMP can inhibit Rap1 activity; either by activating Rap1 GTPase-activating proteins (GAPs, which will convert GTP-loaded Rap1 in an inactive GDP-loaded Rap1), or by inhibiting Rap1 guanine nucleotide exchange factors (GEFs, which will decrease charging Rap1 with GTP). The nature of the Rap1-specific GAP(s) or/and GEF(s) involved in the inhibition of Rap1 in response to cAMP remains elusive [95].
In conclusion, Rap1 seems to be activated in a PKA-dependent and PKA-independent manner on a cell-type specific basis. PKA-dependent activation of Rap1 occurs through direct phosphorylation of SRC by PKA, and subsequent transmission of the signal via Cbl and Crk-L/C3G to Rap1. PKA-independent activation seems to occur through Epac, and may activate specialized subcellular fractions of ERK2.
3.1.7 cAMP/PKA-induced RAF inactivation via PKB or serum-glucocorticoid-inducible kinase (SGK)
PKB was found to act as a potent negative regulator of RAF-1 in myoblast cells and in the breast cancer cell line MCF-7. cAMP elevating agents or PKA can activate PKB in a phosphoinositol-3 kinase (PI3-kinase)-independent, as well as a PI3-kinase-dependent manner, suggesting that PKA can blunt RAF-1 activity through PKB. Similarly, SGK can also inhibit B-RAF kinase signalling and can be activated by elevated cAMP levels in a PKA-requiring manner. Thus, PKB and SGK exemplify an alternative route for cAMP-induced inhibition of RAF-1 and B-RAF, respectively [reviewed in 4].
3.1.8 PKA and Rheb
The small GTPase Rheb interacts with and regulates RAF-1 activity [96]. The interaction of Rheb with Raf-1 is potentiated by growth factors in combination with agents that increase cAMP levels. Protein kinase A-dependent phosphorylation of Ser-43 within the regulatory domain of RAF-1 reciprocally stimulated its interaction with Rheb. Hence, PKA potentiates the activation of ERK through the GTPase Rheb [97].

Figure 4. PKA-regulated MAPK signalling through G-coupled protein receptors. G-protein switching redirects the ?2 adrenergic receptor-ERK signalling. The signalling from the β2 adrenergic receptor is mediated by Gαs, which through adenylyl cyclase (AC) increases cAMP levels and activates PKA. PKA can than modulate the MEK/ERK pathway. Activated PKA can also phosphorylate the β2 adrenergic receptor, which results in uncoupling of Gαs and increased coupling to Gαi. Next, Gαi regulates the MEK/ERK pathway via SRC, RAS, and RAF-1. Alternatively, Epac can activate Rap2B and induce the RAS-RAF-1-MEK-ERK cascade in a Ca2+-dependent manner. The compounds of the signalling pathways that are only used by Gαs are shown in blue, while the transducing proteins solely engaged in the Gαi pathway are represented in light brown. The alternative Rap2B pathway is depicted in orange. Common proteins used by the different pathways are indicated in both colours. This figure is modified after [82].
3.1.9 G protein-coupled receptor and PKA-ERK interaction: PDE-depedent and -independent mechanisms
The G-protein coupled β2-adrenergic receptor forms another mode for cross-talk between PKA and the ERK pathway [reviewed in 82]. Activation of the β2-adrenergic receptor by its agonist results in following successive events (Figure 4): Gαs-protein mediated activation of adenylyl cyclase, generation of cAMP, and activation of PKA. PKA can then stimulate the MEK/ERK pathway through Rap1 and B-RAF. A feed back mechanism by which PKA phosphorylates the β2-adrenergic receptor results in the switch of β2-adrenergic receptor its coupling from Gαs to Gαi with two consequences: (i) decrease in the amplitude of the evoked signal because Gαi inhibits adenylyl cyclase, and (ii) regulation of the ERK1/ERK2 pathway via Gβγ, SRC RAS, and RAF-1. Thus phosphorylation of the β2-adrenergic receptor by PKA diminishes β2-adrenergic receptor coupling to Gαs and increases coupling to Gαi, a process referred to as G-protein switching [Daaka et al., 1997]. The result is an alternative route in the MEK/ERK signalling [Martin, 2004]. Studies by the group of Houslay revealed that PDEs play a major role in G-protein switching and associated ERK activation. Their results are outlined below, but because of the pivotal role of PDE in the PKA-ERK cross-talk, we will first briefly review some structural and functional aspects of PDE.
Intracellular cAMP concentrations are governed by two classes of enzymes with opposite actions. Adenylyl cyclases generate cAMP from ATP [Cooper and Crosswaite, 2006], while cAMP phosphodiesterases (PDE) hydrolyse cAMP into AMP [Houslay et al., 1998] (Figure 1). Eleven families encoding more than 30 different PDE isoforms have been described to date, of which the involvement of the PDE4 family in PKA-MAPK cross-talk has been extensively studied. The four genes that make up the PDE4 family (PDE4A, 4B, 4C, and 4D) are expressed in around 20 different isoforms, which are generated by alternative mRNA splicing or the use of alternative promoters [Houslay et al., 1998; old ref [6]]. The different PDE4 variants are expressed in a cell-specific fashion, and display distinct regulatory properties and intracellular localizations [Houslay and Milligan, 1997; Houslay, 1998; Mehats et al., 2002]. The PDE4 isoforms can be further sub-categorized into the long, short and super-short isoforms. The long variants are characterized by two PDE4-specific upstream conserved regions (UCR1 and UCR2), while the short and super-short isoforms lack UCR1 [Bolger, 1994].
One mechanism by which a cell can establish cross-talk between ERK2 and PKA is through a direct interaction between ERK2 and various PDE4 isoforms. The C-terminal catalytic domain of PDE4B, PDE4C and PDE4D, but not PDE4A can be phosphorylated by ERK2. For example, ERK2 phosphorylates human PDE4D3 at Ser-579 in vitro and in vivo, which decreases the cAMP hydrolyzing activity of PDE4D3 [Hoffmann et al., 1999; Baillie et al., 2000]. Interaction between ERK2 and PDE4 requires the FQF specificity site and the KIM docking sites (see also sections 3.11 and 3.2.2 for KIM docking site). The former is conserved in PDE4A/B/C/D, while the latter motif is not conserved in PDE4A [Mackenzie et al., 2000]. Phosphorylation of the long PDE4B, PDE4C, and PDE4D variants leads to inhibition of the phosphodiesterase activity and hence increased cAMP levels, while of the short PDE4B2 form was activated upon phosphorylation by ERK2 [Baillie et al., 2000]. Activated PKA can phosphorylate the long isoforms at their N-terminal domain (e.g. Ser-13 and Ser-54 for PDE4D3), a modification that ablates the inhibitory effect of ERK2. Thus the activation of ERK2 inhibits or stimulates cAMP/PKA signalling depending on the PDE4 subtypes, which are expressed in a cell-specific manner. Additional levels control the outcome of phosphorylation of PDE4 by ERK and PKA. Oxidative stress induces phosphorylation of human PDE4D3 at Ser-239 by an unknown kinase and at Ser-579 by ERK2. Phosphorylation of Ser-239 switched the effect of phosphorylation by ERK at Ser579 from inhibition to activation [Hill et al., 2006]. Interestingly, ERK2 activation in smooth muscle cells has been shown to reduce the stability of the super-short PDE4D1 transcripts. Prolonged elevation of cAMP resulted in PKA-dependent induction of mRNA levels PDE4D1 and PDE4D2, while simultaneous activation of both the PKA and MEK-ERK signalling cascades blunted this cAMP-mediated increase in PDE4D transcript levels [139]. In conclusion, the effect of ERK2 on the activity of PDE4 in a particular cell depends on the expression levels and pattern of the PDE4 isoforms, as well as additional phosphorylations of PDE4 in response to stimuli. This complex regulation of PDE4 activity allows tailoring a cell-type specific cross-talk between the cAMP and ERK pathways [127, reviewed in 140]. The ERK-PDE4-PKA connection was shown to play an important role in memory processes [Zhang et al., 2004] as activation of ERK signalling enhances long-term memory [Purcell et al., 2003], while inhibition of cAMP/PKA impairs long-term activity [Koh et al., 2002]. Abrogation of PDE4 activity by ERK2, accompanied by elevated cAMP levels is also an essential mechanism to overcome inhibition of regeneration after nerve injury [Gao et al., 2003].
Houslay and his team went on to show that the PDE-mediated link between PKA and ERK2 involves another group of proteins: the β-arrestins. β-arrestins are ubiquitously expressed intracellular adaptor and scaffolding proteins that carry out several important functions in the cell. Their best-know role is in G protein-dependent signaling pathways. Activated G protein-coupled receptors (GPCR such as the β2-adrenergic receptor) are rapidly phosphorylated by specialized enzymes called G protein-coupled receptor kinases. Phosphorylated receptors bind the multifunctional adaptor proteins β-arrestin with high affinity. β-arrestin binding blocks further G protein coupling, leading to desensitization of G protein-dependent signaling pathways [for recent reviews see DeFea, 2007; DeWire et al., 2007; Pfleger et al., 2007; Violin and Lefkowitz, 2007]. β-arrestin are also implicated in transcription, apoptosis, development, cellular migration, and metastasis [Buchanan and DuBois, 2006; Ma and Pei, 2007].
β-arrestins have the capacity to scaffold PDE4 isoforms from all the four subfamilies [Perry et al., 2002; Bolger et al., 2003], and this can serve to regulate the switching of coupling the β2-adrenergic receptor from Gs to Gi and hence activation of ERK. Indeed, activation of the β2-adrenergic receptor in cardiac myocytes or HEK293 cells overexpressing this receptor resulted in recruitment of β-arrestins, as well as the long isoforms of PDE4D3 and PDE4D5, and was accompanied by Gs to Gi switching. Inhibiting PDE4 activity by the selective inhibitor rolipram or by overexpression of a catalytically inactive PDE4D5 mutant markedly increased PKA-dependent phosphorylation of the β2-adrenergic receptor and potentiated β2-adrenergic receptor-induced activation of ERK. Thus β-arrestin-mediated recruitment of PDE4 regulates the PKA phosphorylation status of the β2-adrenergic receptor and Gs to Gi switching (see above), and enhances ERK activation [Baillie et al, 2003]. RNA interference studies allowing PDE4 isoform-selective depletion confirmed that β-arrestin-mediated delivery of PDE4D5 plays a major role in regulating β2-adrenergic receptor switching and activation of ERK [Lynch et al., 2005; Willoughby et al., 2007]. PDE4D5 can also bind to the scaffold protein RACK1 (receptor for activated C-kinase), and β-arrestins and RACK1 bind PDE4D5 in an exclusive manner [Bolger et al., 2006; Smith et al., 2007]. Si-RNA-mediated knockdown of RACK1 increased the levels of PDE4D5 associated with β-arrestins and attenuated PKA phosphorylation of the β2-adrenergic receptor and ERK activation. Thus the ratio of RACK1 and β-arrestins protein levels may affect the outcome of PDE4D5-PKA-ERK pathway in a cell-specific manner [Bolger et al., 2006].
Keiper and co-workers described yet another route by which GPCR activate ERK1/2 in a cAMP-dependent manner [83]. By overexpressing of dominant negative variants, and employing specific chemical inhibitors and the specific Epac activator 8-pCPT-2Me-cAMP, they found that ERK1/2 phosphorylation in HEK293 cells that ectopically expressed the β2-adrenergic receptor in response to forskolin or adrenaline exposure was PKA-, SRC-, Gi-, and Rap1A-independent, but required cAMP-mediated Epac1 activation, followed by Rap2B activation. Rap2B then activated PLC-ε, which led to increase in intracellular Ca2+ concentrations and activation of the Ca2+-regulated GEF, RasGRF1 (=CalDAG-GEF II). RasGRF1 subsequently activated RAS, which next led to ERK1/2 phosphorylation via RAF-1 and MEK1/2 (Figure 4). A similar mechanism was operational for PGE2-induced ERK1/2 activation in the N1E-115 neuroblastoma cells [83].
In conclusion, four pathways that link cAMP to ERK1/2 phosphorylation upon activation of GPCR have been identified: the PKA-dependent pathway acts either via SRC>Rap1>B-RAF>MEK1/2>ERK1/2 or through PKA-mediated G-protein switching and Gβγ-induced activation of RAS>RAF-1>MEK1/2>ERK1/2. The PKA-independent pathways may use cAMP-triggered Epac1 activation and subsequent Rap1, B-RAF, and MEK1/2 to activate ERK1/2, or cAMP/Epac1-provoked activation of the cascade Rap2B>PLC-ε>Ca2+>RasGRP1>RAS>RAF1>MEK1/2.
3.1.10 3.1.10. Receptor tyrosine kinase-cAMP/PKA interaction
PKA can also stimulate the RAS/RAF/MEK/ERK pathway by targeting the epidermal growth factor receptor (EGFR). Studies in human non-transformed breast epithelial MCF-10A cells had demonstrated that overexpression of RI? enabled these cells to grow in the absence of serum, while selective downregulation of RIα expression reduced cells to enter the S phase. Furthermore, MCF-10A stably transformed with RIα displayed constitutively active ERK1/2. It was therefore no complete surprise to find that type I PKA and the activated EGF receptor (EGFR) interacted, an association which is probably mediated by the direct binding of RIα to the adaptor Grb2 [84]. These findings suggest that RIα can mediate mitogenic signalling via Grb2-mediated recruitment of type I PKA to the EGFR and subsequent transmission by the RAS/RAF/MEK/ERK pathway.
Studies with cardiac myocytes had shown that EGF not only activated ERK1/2, but also resulted in cAMP accumulation and PKA activation [85]. The interconnection between the two pathways originates at the level of the receptor: ERK activation occurs through the classical RAS-RAF-MEK cascade, while the tyrosine kinase activity of the EGF receptor phosphorylates the α-subunit of Gs. This augmented Gsα its ability to stimulate adenylyl cyclase and hence to generate cAMP. The exact phosphorylation sites and the mechanism by which this modification activates Gsα remain unclear. Simultaneous activation of the ERK and PKA pathways by the EGF receptor may allow the amplification and/or endurance of the signal and hence result in multiple cellular responses. The increase in cAMP levels augments the beating rate and contractility in intact hearts, indicating that, under circumstances when optimal heart functions are required, signals may accomplish this by engaging several signalling pathways [85].
3.1.11 3.1.11. cAMP/PKA-protein tyrosine phosphatase
The cAMP/PKA pathway may integrate indirectly with the MAPK pathway through the use of protein tyrosine phosphatises (PTP) that control the phosphorylation levels of MAPKs [Pulido et al., 1998]. To dephosphorylate ERK, PTP needs to dock onto ERK through its kinase interaction motif (KIM). cAMP may activate PKA, which in turn may phosphorylate a serine residue within the KIM domain. This abolishes binding of PTP to ERK and hence dephosphorylation, and ultimately in activation of ERK1/2 [86; old [98]]. cAMP-induced activation of ERK1/2 through PKA-mediated inactivation of PTP occurs also in rat granulose cells after follicle-stimulating hormone (FSH) treatment. This activation of ERK1/2 was PKA-dependent because myristoylated protein kinase inhibitor (Myr-PKI) abrogated FSH-induced ERK1/2 activation. FSH-induced ERK1/2 activation was also inhibited by the MEK inhibitor PD98059. FSH did not, however, increase the phosphorylation levels of MEK1/2, nor did it enhance RAS and RAF-1 activity, suggesting that PKA-mediated activation of ERK occurred downstream of RAS/RAF. FSH-induced ERK1/2 activation was accomplished by PKA-triggered phosphorylation of a 100 kDa PTP. PKA-mediated phosphorylation of the PTP resulted in dissociation of PTP from ERK1/2 and sustained phosphorylation of ERK1/2 [reviewed in 87]. The restricted expression pattern of PTP and PTP-SL implicates that this level of PKA-directed MAPK inactivation is highly cell-specific. PTPs can also dephosphorylate p38 MAPK and JNK (see 3.2. and 3.3.), so that this cross-talk mechanism is not unique for PKA and ERK1/2.
3.1.12 Effect of the MEK/ERK MAPK pathway on PKA activity through an autocrine loop
Most examples discussed so far illustrate the effect of cAMP/PKA on the RAF/MEK/ERK pathway. Reversely, this MAPK cascade can also modulate PKA activity. One mechanism is PKA activation through ERK2-mediated inhibition of PDE (see section 3.7.). Another mode of ERK-driven PKA activation was described in human arterial smooth muscle cells. Treatment of these cells with platelet-derived growth factor rapidly stimulated cAMP synthesis through a mechanism requiring intracellular Ca2+, PKC activity, and MEK/ERK activity. MAPK-induced cAMP accumulation and PKA activation was accomplished through ERK-mediated activation of cytosolic phospholipase A2, release of arachidonic acid, prostaglandin PGE2 synthesis and secretion, and subsequent activation of its receptor. This G-protein coupled receptor induced adenylyl cyclase through GTP-loaded Gα upon ligand binding, which resulted in cAMP generation and PKA activation [47,88].
Another mechanism by which a component of the MAPK pathway affects PKA is exemplified by the RAF-1-mediated activation of adenylate cyclases, a family of enzymes convert ATP into cAMP. Currently, 10 different mammalian isoforms, including nine membrane-bound and one soluble variant, have been described [Cooper, 2003]. RAF-1 has been shown to phosphorylate and activate adenylate cyclases type II, V, and VI, but not I [Ding et al., 2004; Beazely et al., 2005]. Thus, Raf-1-mediated activation of adenylate cyclase enhances intracellular cAMP levels and can lead to PKA activation. On the other hand, PKA can inactivate adenylate cyclase isoforms through phosphorylation [Beazely et al., 2005], so that fine-tuned mechanisms seem to determine the on (through RAF-1)/off (through PKA) status of adenylate cyclases.
3.2 cAMP/PKA and p38 MAPK
As for the MEK/ERK pathway, cAMP/PKA seems to have opposite effects on p38 MAPK activity depending on the cell type examined. But in contrast to ERK2 which could influence PKA activity through activation or inhibition of PDE, no effect of the p38 MAPK module on PKA has been reported, so far.
Several studies have reported PKA-dependent activation of p38 MAPK in a variety of cells, including NIH3T3, macrophages, SK-N-MC, PC12, MC3T3-E1, CHO, colon cancer cells, adipocytes, rat primary granulosa cells, and adult mouse cardiomyocytes. However, forskolin has also been shown to inhibit p38 MAP kinase activation in e.g. HUV-EC cells and in thymocytes [74,87,89]. Currently, several different mechanisms by which cAMP/PKA can interfere with the p38 MAPK module have been described. PKA can influence the activity of the p38 MAPK cascade through: (i) Rap1-mediated regulation, (ii) PTPs acting on p38 MAPK, (iii) protein phosphatases targeting upstream activators of p38 MAPK, (iv) glia maturation factor, (v) the p160ROCK kinase, or (vi) the small GTP-ase Rit. These different mechanisms will be briefly commented here.
3.2.1 Rap1-mediated regulation of p38 MAPK
As elaborated in section 3.1., Rap1 plays a central role in PKA-ruled regulation of the MEK/ERK pathway, a function that may be extended to the modulation of p38 MAPK activation in cerebellar granule cells. Exposure of these cells to cAMP analogues induced big K+ channels opening and p38 MAPK, but not ERK1/2 or JNK phosphorylation in a PKA-independent, but Epac-Rap-dependent manner. The precise mechanism by which Epac/Rap mediates p38 MAPK activation remains unexplored [90]. Another study documented the role of Rap1, but not Epac in the activation of p38 MAPK [old references 91,92]. This investigation demonstrated that adrenaline-induced glucose uptake in rat vascular smooth muscle cells was mediated through the β-adrenergic receptor-Gs-adenylyl cyclase-cAMP cascade. However, neither PKA nor Epac acted as cAMP acceptor since inhibition of PKA or specific activation of Epac with 8-pCPT-2Me-cAMP did not affect adrenaline-induced glucose uptake. However depletion of Rap1 by siRNA strongly reduced glucose uptake, as did the p38 MAPK inhibitor SB203580 [91,92]. Further studies are required to examine the role of PKA and Epac in this process and to elucidate the mechanism by which Rap1 triggers activation.
Rap1 has also been shown to activate p38 MAPK through MKK3/6 in the hippocampus after exposure to the metatropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine [93], and after stretching of L-929 mouse fibroblasts [94], but it is unclear how exactly the Rap1 signal is transmitted to p38 MAPK.
3.2.2 Protein tyrosine phosphatases acting on p38 MAPK
Another mode to regulate the activity of MAPK is to prevent their dephosphorylation (see also 3.1.11). The haematopoietic protein tyrosine phosphatase (HePTP, also known as leukocyte protein tyrosine phosphatase or LC-PTP), PTP-SL, and STEP have emerged as major regulators of MAPK functions on the basis of their association with ERK1/2 and p38 MAPK through a 16-amino acid kinase interaction motif (KIM) [95,96]. HePTP binds p38 MAPK and mediates dephosphorylation and hence inactivation of p38 MAPK. In T lymphocytes it was shown that PKA phosphorylates HePTP at Ser-23. Phosphorylation of Ser-23 by PKA inhibits the negative regulatory function of HePTP on p38 MAPK and ERK2, while PP1 seems to dephosphorylate this site and positively regulates the phosphatase activity of this enzyme [86,97]. A similar mechanism seems to be operational for PTP-SL. PTP-SL associates with ERK2 and p38α MAPK through KIM and retrains these MAPK in the cytoplasm. Moreover, the tyrosine phosphatase activity keeps the MAPK in an inactive state. When PKA phosphorylates PTP-SL at serine-231, it impairs PTP-SL its enzymatic activity and its ability to bind and dephosphorylate ERK2 and p38?. The KIM sequences required for docking ERK2 and p38α are conserved in STEP. As PKA can also phosphorylate STEP in vitro and can interact with ERK1/2, suggests that STEP may also be a target for the interaction between PKA and MAPK signalling [95,98], Thus PKA can indirectly modulate the phosphorylation/activation state of ERK1/2 and p38 MAPK through phosphorylation-mediated inhibition of PTPs that control the subcellular localization and catalytic activity of these MAPKs.
3.2.3 Protein phosphatases targeting upstream activators of p38 MAPK
Another mechanism for PKA-induced p38 MAPK activation involves protein phosphatases that regulate the activity of upstream activators of p38 MAPK. Stimulation of β-adrenergic receptors in brown adipocytes resulted in elevated cAMP levels, and PKA and p38? MAPK activation. Since cAMP can activate Rap1, and Rap1 can promote p38 MAK activation [94,99], cAMP-induced activation of p38? MAPK may therefore occur through Rap1. However, in these cells, Rap1 was not activated and PKA was the only mediator of β-agonist induced p38 MAPK activation. This p38 MAPK activation seems to be mediated by its upstream activator MKK3. Indeed, siRNA studies showed that depletion of MKK3, but not MKK6, completely abolished β-adrenergic receptor-induced p38α MAPK activation [100]. A possible way for such a PKA-MKK3 interconnection may stem from PKA-mediated inactivation of protein phosphatases. For example, the protein phosphatases PP2Cα, PP2Cβ-1, and PP2Cε negatively regulate the upstream activators of p38 MAPK, TAK1 or MKK3/6 [Hanada et al., 2001], and it is possible that PKA inactivates PP2C, which sustains activation of the TAK/MKK3/6/p38 MAPK pathway. So far, no role for PP2C has been attributed in PKA-mediated activation of TAK and /or MKK3/6. An alternative model involves the TAK1-interacting proteins TAB. TAK1 resides in an active complex with TAB1 and TAB2 or TAB3, which is activated upon phosphorylation [101]. Whether TAB-TAK1 complexes are substrates for PKA is unknown, but in this scenario, PKA would render TAK1 in an activator of the p38 MAPK cascade.
3.2.4 Glia maturation factor mediates PKA-induced p38 MAPK activation
An unexpected mechanism to control p38 MAPK activity in a PKA-dependent manner emerged from the studies by Lim and co-workers [old 103-105]. Glia maturation factor (GMF) is a 17 kDa protein that may play a role in cell growth and differentiation, and that regulates the expression of cytokines and chemokines. Moreover, studies with GMF deficient mice have assigned a role for this peptide in motor performance and learning [102]. PKA, PKC, CKII, and RSK can all phosphorylate GMF in vitro. Furthermore, GMF interacts with p38 MAPK in vivo and PKA-phosphorylated GMF, but not GMF phosphorylated by PKC, CKII or RSK or unphosphorylated GMF inhibited ERK activity in vitro, while the in vitro kinase activity of p38 MAPK was strongly enhanced. Overexpression of GMF in PC12 cells and primary astrocytes resulted in augmented p38 MAPK activity [103-105]. The mechanism and biological consequences of PKA-induced phosphorylation of GMF and its effect on ERK1/2 and p38 MAPK signalling remain unclear. These data demonstrate that the same substrate for PKA, GMF, may exert opposite effects on two different MAPK cascades.
3.2.5 PKA-induced activation of p38 MAPK through p160ROCK
Studies on tumour cell motility performed by the laboratory of Reshkin disentangled still another mechanism by which PKA may accomplish p38 MAPK activity [106,107]. This group showed that hypoxia or serum-deprivation, a common tumour microenvironmental condition, activated the Na+/H+ exchange isoform (NHE1), which is the main regulator of intracellular and extracellular pH and exerts a fundamental role in tumour cell motility. Low serum-induced NHE1 activity was orchestrated by PKA, which by means of direct phosphorylation of RhoA at Ser-188 inhibits the ser/thr kinase p160ROCK. Decreased activity of p160ROCK resulted in reduced p38α MAPK activity and increased NHE1 activity. The PKA>RhoA>p160ROCK>p38 MAPK cascade defines a novel level of PKA-MAPK interaction, but the fine mechanisms by which p160ROCK modulated p38α MAPK needs to be resolved [106,107].
3.2.6 cAMP-Rit-p38 MAPK pathway
Recently, a novel cAMP-dependent, PKA-independent pathway to activate p38 MAPK was described by Shi and co-workers [old 108,109]. They showed that binding of pituitary adenylate cyclase-activating polypeptide 38 (PACAP38) to its adenylyl cyclase-coupled receptor resulted in increase of intracellular cAMP levels and activation of the small GTPase Rit in PC6 cells. The activation of Rit was PKA-independent, and Epac-dependent, but neither Epac1 nor Epac2 activated Rit directly. Activated Rit did activate the p38 MAPK isoforms α and γ, but not δ (β was not tested). Furthermore, Rit had no obvious effect on ERK1/2 in these cells, although it could activate MEK/ERK under other experimental conditions or in other cells [108,109]. The mechanisms by which Epac induces Rit, and by which Rit regulates specific isoforms of p38 MAPK are still unknown.
3.3 cAMP/PKA and JNK
The effect of cAMP on the other MAPK signalling cascades has been less explored, but shortly after the discovery of the modulation of the RAF/MEK/ERK by cAMP, it was demonstrated that 12-O-tetradecanoyl phorbol 13-acetate (TPA)-, concanavalin-, and IL-2-induced activation of JNK1/2 was inhibited by cAMP in splenic T lymphocytes and T lymphoma cell lines, while cAMP only poorly antagonized ERK1/2 activity [110]. Similarly, TPA- and ionomycin-induced JNK activation was hampered by forskolin in T cells [111]. In a recent study, Black and co-workers demonstrated that forskolin greatly reduced TGFβ1-induced JNK, p38 MAPK and ERK1/2 phosphorylation in a cAMP/PKA-dependent manner in human lung (IMR90) fibroblasts. In gingival fibroblasts, however, forskolin only inhibited TGF?1-triggered phosphorylation of JNK, but had little effect on the phosphorylation of ERK1/2 or p38 MAPK [112]. Inhibition of JNK activity by increased cAMP levels has also be observed in other cells such as acute lymphoblastic leukaemia cells and cardiac muscle cells [113,114]. The PKA inhibitor KT5720 also reversed the inhibitory effect of forskolin or cAMP analogues on JNK activation in cardiac muscle cells, suggesting a PKA-dependent mechanism [113]. The molecular mechanism underlying the inhibition of JNK by cAMP/PKA remains unclear, but Hsueh and Lai observed that suppression of JNK activity by cAMP required de novo RNA and protein synthesis, suggesting the involvement of newly synthesized inhibitor(s) [old 110]. This may also explain why ablation of JNK activity by cAMP is delayed compared to ERK inactivation. Indeed, effects of increased cAMP levels on ERK activity are usually monitored within minutes, while inhibition of JNK is relatively slow (hours). The nature of this inhibitor remains elusive, but the MAPK phosphatase MKP-1 might be a strong candidate because its expression inclines in cells exposed to cAMP-elevating agents [115,116], and JNK is a genuine substrate of MKP-1 [reviewed in 117]. Thus through cAMP-induced upregulation of MKP-1 expression and subsequent dephosphorylation of active JNK, cAMP has the potential to inhibit JNK activity.
Studies with vasoactive intestinal peptide and the pituitary adenylate cyclase-activating polypeptide may provide insight into another mechanism by which cAMP affects JNK activity. These two neuropeptides stimulate the generation of cAMP and inhibited JNK activity in microglia cells. This reduction in JNK activity could be mediated by PKA and occurred at the level of MEKK1 activity, the upstream activator of the MEK4/JNK pathway [118]. PKA did not phosphorylate MEKK1 in vitro, but TAK, which lies upstream of MEK4/7 (see Figure 2), was a good in vitro substrate for PKA [119]. The putative role of TAK in the crosstalk between cAMP/PKA and JNK pathways in cells needs to be further explored.
The activity of JNK may also be regulated by cAMP, as JNK activation was ablated in the presence of the PKA inhibitor H89 in breast cancer cells [120]. In adjacent breast adipose fibroblasts either specific activation of PKA or Epac with cAMP analogues stimulated both JNK and p38 activity [121]. To date, two mechanisms for cAMP/PKA-induced activation of the JNK pathway have been suggested. One scenario implies the tyrosine kinase receptor RET. Phosphorylation of RET at Tyr-1062 is crucial for activation of major intracellular signalling pathways, including the RAS/ERK, PI3K/AKT, JNK, p38 MAPK, and ERK5 pathways. The RET-JNK pathway seems to proceed through DOK1>NCK>the small GTPases Rho/Rac/Cdc24>PAK>MEKK>MEK4/7>JNK [reviewed in 122]. PKA can interfere with the RET-JNK pathway via direct phosphorylation of Ser-697 RET, a modification that promotes the JNK signalling pathway [123]. The exact molecular mechanism by which phosphoSer-697 RET affects the JNK pathway has not been unravelled. An alternative manner by which cAMP arouses JNK activity involves Epac. Overexpression of Epac in HEK293T cells could activate JNK1 independently of Rap1 [old 124]. Whether cAMP binding to endogenous Epac affected activation of JNK1 was not tested, but cAMP-elevating agents did not alter JNK1 activity induced by ectopic expressed Epac. The authors did not investigate whether Epac interacts directly with JNK. Moreover, the exact mechanism by which Epac increases JNK-1 remains to be solved, but the RAS-exchange domain (REM domain) is absolutely required [124]. Mochizuki and colleagues could not confirm the findings by Hochbaum et al., since they failed to detect JNK activation in HEK293T cells overexpressing Epac [125]. Thus the putative role of Epac in JNK signalling in HEK293 cells remains controversial. The use of the RNA interference and the specific Epac activator 8-pCPT-2-Me-cAMP may enable to establish the participation of Epac in the cross-talk between cAMP and the JNK pathways. Moreover, the role of Epac in JNK signalling may be cell-specific because stimulation of endogenous Epac in Jurkat T cells with 8-pCPT-2-Me-cAMP exerted an inhibitory effect on c-JUN-mediated transcription, suggesting that Epac may negatively regulate the JNK pathway [126].
3.4 PKA and MEK5/ERK5
Dodge-Kafka and co-workers reported that the scaffold protein muscle A kinase-anchoring protein (mAKAP) gathers several "players" of the PKA and MAPK "teams", including Epac1, PKA, the PDE isoform PDE4D3, and ERK5, and that this intimate clustering of these proteins orchestrates multiple interconnections between the pathways [old 127,128]. mAKAP-associated PDE4D3 binds directly to ERK5, allowing ERK5 to phosphorylate PDE4D3. This results in decreased phosphodiesterase activity accompanied by increased cAMP levels and PKA activity. Thus ERK5 can influence the activity of PKA by diminishing the enzymatic activity of PDE4D3. A third cAMP-MAPK interaction that occurs at mAKAP is cAMP modulation of ERK5 activity. cAMP inhibits ERK5 activity in a PKA-independent manner through Epac, which is recruited by PDE4D3 to the mAKAP complex. All these findings illustrate how mAKAP coordinates multiple interactions between cAMP and MAPKs. Activation of ERK5 and the resulting phosphorylation of PDE4D3 results in increased cAMP and the activation of PKA. PKA can then phosphorylate (activate) PDE4D3, which causes a negative feedback on PKA due to reduced cAMP levels. Sustained elevated cAMP concentrations will allow Epac-interceded activation of Rap1, resulting in inhibition of ERK5, which is recruited to mAKAP by PDE4D3. Because PKA has higher affinity for cAMP than Epac, Epac might only take effect at higher cAMP levels. The mechanisms by which Rap1 hampers ERK5 activity was not addressed [127,128].
Independent studies by the group of Cobb confirmed these findings and elucidated the mechanism for cAMP-mediated inhibition of ERK5 [old 129,130]. ERK5 was activated by cAMP-elevating agents in growing NIH 3T3 and PC12 cells, and in C2C12 myoblasts. However, in confluent, serum-starved cells, elevation of the cAMP levels abrogated activation of EGF-induced (HeLa and C2C12 cells) and FGF-induced (NIH 3T3 cells) ERK5 activation. Moreover, higher concentrations of EGF overrode the inhibitory effect of cAMP on ERK5 activation [old 129]. These results demonstrate a negative regulation of ERK5 by cAMP under certain cellular contexts. cAMP also reduced the activity of the ERK5 kinase, MEK5, and of its upstream activator MEKK2 (Figure 2). Both ERK5 and MEKK2 inhibition was found to be PKA-dependent. PKA phosphorylated MEKK2 on Ser-153 in vitro and in vivo, but did not reduce the in vitro kinase activity of activated MEKK2 isolated from growth factor-treated cells. The authors observed that once ERK5 is activated in cells, it is refractory to inhibition by cAMP. Likewise, activated MEKK2 was resistant to cAMP inhibition. This made the authors propose that phosphorylation of MEKK2 by PKA breaks the connection of MEKK2 with its upstream regulatory mechanism, thereby interrupting activation of MEK5 and ERK5. However, once MEKK2 is activated, PKA-mediated phosphorylation of MEKK2 cannot interrupt the MEK5-ERK5 activation cascade [old 130]. The biological importance of cAMP-triggered inhibition of the MEKK2/MEK5/ERK5 pathway correlates with prevention of growth factor-induced proliferation. When the cAMP concentration is low, growth factors activate ERK5, leading to proliferation. When the concentration of cAMP is high, growth factors lose the ability to stimulate ERK5, but may display increased cell survival. Consistent with this hypothesis is the observation that confluent cells treated with growth factors in the presence or absence of cAMP elevating agents demonstrated that fewer of the growth factor-treated cells with increased cAMP levels cells underwent apoptosis compared to cells treated with growth factors alone [129,130].
3.5 PKA and RSK
A recent study described in vivo interaction between inactive (dephosphorylated) RSK1 and the regulatory RI subunit of PKA, while active (phosphorylated) RSK1 preferentially bound the catalytic C-subunit of PKA [old 131 and reviewed in Houslay, 2006]. The binding of inactive RSK1 to RI decreased the interaction between the regulatory and catalytic subunits of PKA, while association of active RSK1 with the C subunits enhanced the interaction between the R and C subunits. This promoted the formation of inactive PKA holoenzyme, and thus active RSK1 reduced the ability of cAMP to activate PKA. The regions of RSK1 and the PKA subunits involved in the complex formation have not yet been identified, but the closely related RSK2 and RSK3 were unable to interact with PKA. This may suggest that domains with less homology between the RSK family members may be involved in the binding of PKA. The interaction between RSK1 and PKA illustrates a novel mechanism for the regulation of PKA: activated RSK1 could inhibit PKA by strengthening the assembly of the R- and C-subunits. The biological importance of the PKA-RSK1 interaction may be dual because PKA-RSK1 cross-talk not only modulated the activity of PKA, but also permitted the regulation of the spatial localization of RSK1, which may help to regulate of the biological actions of RSK1. Indeed, association of active RSK1 with PKA apparently ensured its nuclear localization and therefore positioned RSK1 in the proximity of its substrates. RSK1 targets both nuclear substrates (e.g. histone H3, the spindle checkpoint regulatory protein Bub1, and the transcription factors CREB and c-Fos) and cytosolic proteins (e.g. the pro-apoptotic protein BAD and the tumour suppressor tuberous sclerosis complex-2, TSC2, also known as tuberin), allowing PKA to interfere with RSK1-mediated phosphorylation of these substrates [131 and references therein]. ERK1/2 can phosphorylate TSC2 at Ser-540 and Ser-664, while RSK-1 phosphorylates tuberin at Ser-1798. These post-translational modifications inhibit the function of TSC2 [132,133]. Because TSC-2 has been shown to function as a negative regulator of Rap1 [134], a feed back mechanism of Rap1-induced activation of the MEK>ERK>RSK pathway mediated by RSK-1-catalyzed phosphorylation of TSC-2 can be imagined.
The upstream activator of RSK1, ERK2 may exert opposite functions on PKA: ERK2-mediated phosphorylation of RSK1 will allow pRSK1 to bind the catalytic subunits of PKA and reduce the ability of cAMP to induce activation of PKA. On the other hand, ERK2 can phosphorylate and thereby inhibit PDE4 long variant, resulting in increased cAMP concentrations and PKA activation (see section 3.7.). Thus ERK2 can inhibit PKA through activation of RSK1, while it can activate PKA through inhibition of PDE4.
In the T84 intestinal epithelial cell line, cAMP activated ERK1/2, through PKA type II and not PKA type I [135]. Thus PKA type II may modulate PKA type I via the ERK2-RSK1-PKA type I interaction discussed above.
3.6 PKA and MK5
MK5 (or its human homologue PRAK) was originally described as a down-stream protein kinase of p38 MAPK. MK5 seems to be ubiquitously expressed in all vertebrates examined so far, but is absent in Caenorhabditis elegans and Drosophila [reviewed in Gaestel, 2006]. The biological functions of MK5 are poorly understood, but studies with knock-out mice and knock-in mice have shed some light on the functions of MK5. Mice deficient in MK5 are fertile and develop normally, but where more susceptible to chemical-induced skin cancer than their control littermates. Moreover, in vitro studies with MK5-/- cells revealed a role for MK5 as tumour suppressor [Sun et al., 2007]. On the other hand, mice expressing a constitutive active MK5 mutant demonstrated anxiety-related traits and locomotor differences compared to their control littermates, which may implicate a role for MK5 in neurological processes [Gerits et al., 2007a]. Moreover, we have shown that MK5 is involved in PKA-induced microfilament rearrangement [Gerits et al., 2007b= old 137]. We have recently identified a novel interaction between the PKA and MAPK pathways [old 137]. We showed that the catalytic subunit of PKA (Cα) interacts with MK5, but not with the closely-related MK2 in vivo, and that C? can induce nuclear export of MK5, an event that requires the kinase activity of both proteins. Cα also increased the phosphorylation pattern of MK5 and enhanced the catalytic activity of MK5. Nuclear export of MK5 required Cα to enter the nucleus, and mutations in potential PKA phosphorylation sites of MK5 also affected the nuclear-cytoplasmic redistribution of MK5. The regions of MK5 and of Cα required for the interaction have not been mapped, nor has it been solved whether Cα actually acts as a carrier to transport MK5 out of the nucleus or whether PKA-mediated phosphorylation is sufficient to cause a conformation change in MK5, thereby unmasking its nuclear export signal, as has been shown for p38 MAPK [136]. However, overexpression of Cα tagged with a nuclear localization signal (NLS) showed exclusive nuclear distribution of the Cα-NLS fusion protein, while MK5 was relocated to the cytoplasm. This may indicate that MK5 is not exported out of the nucleus in complex with Cα. We further went on to demonstrate that depletion of MK5 interfered with cAMP-provoked F-actin rearrangements. Our findings point to a scenario where PKA redirects a cytosolic distribution of activated MK5, which enables MK5 to participate in F-actin remodelling by phosphorylation of Hsp25/27 [137, S.K., unpublished results]. MK5 has also been shown to activate p53 by direct phosphorylation [138]. It is not known whether PKA can influence MK5-mediated p53 phosphorylation, but because PKA can activate MK5, it may link PKA to a vital protein with crucial central functions in the cells.
3.7 Common substrates for PKA and MAPK
The PKA and MAPK may converge to the same substrates and phosphorylate identical or different phosphoacceptor sites. This may affect the activity of the substrate mediated by either one of the pathways. Some examples of common PKA/MAPK substrates and their result of targeting the same substrate are given below.
Residue Ser-133 in the transcription factor cAMP response element-binding protein (CREB) acts as the phosphoacceptor site for many protein kinases, including PKA and the MAPKs RSK, MSK1, MSK2, p70SK6, MK2 and MK3 [reviewed in 141]. Phosphorylation of CREB at Ser-133 by multiple protein kinases allows modulating the kinetics and stoichiometry of CREB phosphorylation. Indeed, forskolin-induced phosphorylation of CREB at Ser-133 endured beyond the time-point when PKA becomes refractory because the incoming signal was transmitted from PKA to p38 MAPK and subsequently MSK-1, which sustained Ser-133 phosphorylation [Delghandi et al., 2005]. MSK1 and PKA also cooperate to activate CREB in a subset of hippocampal CA1 pyramidal neurons [142].
The pro-apoptotic BAD protein forms another protein that can be phosphorylated by both PKA and MAPKs. BAD can dimerize with the anti-apoptotic proteins BclX and Bcl-2, which results in neutralization of their anti-apoptotic activity. However BAD phosphorylated at Ser-122 by PKA or/and RSK1 will bind 14-3-3, this will release BclX and Bcl-2 and promote cell survival [143]. The concerted action of PKA and RSK1 may enforce the protection mechanisms of cells against apoptosis. The MEK5/ERK5 MAPK cascade also converges to BAD. Ectopic expression of constitutive active MEK5 in bovine lung microvascular endothelial cells induced BAD phosphorylation Ser112 and Ser136 and increased the anti-apoptotic activity of BAD [144].
Phosphorylation-mediated regulation of protein activity by a certain protein kinase may be counteracted by another kinase. The transcription factor ER81 is phosphorylated in vivo by RSK1 on Ser-191 and Ser-216, and this stimulated the transcriptional activity of ER81. Similar to RSK1, PKA phosphorylates ER81 on the same sites, but additionally, PKA targets ER81 on Ser-334. However, phosphorylation of Ser-334 severely reduced the DNA-binding ability of ER81 but still enhanced its transactivation potential. The authors suggested that PKA-mediated phosphorylation favours transcription of promoters with high but not with low ER81 affinity binding sites [145].
As outlined in section 3.7., PKA and ERK2 can both phosphorylate the long variant of PDE4 isoforms, however with opposite effects. Phosphorylation of PDE4 in the catalytic domain by ERK2 inhibited the cAMP hydrolyzing activity of PDE4, while phosphorylation of the N-terminal region of PDE4 by PKA ablated the inhibitory effect of ERK2-mediated phosphorylation.
In vitro studies demonstrated that tyrosine hydroxylase, an enzyme involved in catecholamine biosynthesis, was phosphorylated by the MAPKAPKs MSK1 at Ser-40 and MK5 on Ser-19. Phosphorylation of both sites augmented the affinity for 14-3-3, but only the interaction with Ser-19 increased tyrosine hydroxylase activity. The phosphorylation of tyrosine hydroxylase on Ser-19 caused a threefold increase in the rate of phosphorylation of Ser-40 [146]. PKA phosphorylates Ser-40 in vivo, a modification that is essential for activation of tyrosine hydroxylase by PKA [147]. It remains to be established whether tyrosine hydroxylase is a genuine MSK1 and MK5 target, but arsenite exposure rapidly increased Ser19 phosphorylation, concomitant with phosphorylation of the MK5 activator p38 MAPK. These findings underscore the assumption that MK5 may phosphorylate tyrosine hydroxylase at this site [148]. The biological implications of dual phosphorylation of tyrosine hydroxylase by MAPKs and PKA have not been examined.
Peroxisome proliferator-activated receptors (PPARα, β, and γ) are transcription factors whose activity is regulated by multiple phosphorylation events. ERK2 phosphorylates PPARα on Ser-12 and Ser-21 in vitro, while Ser-6, Ser-12, and Ser-21 are p38 MAPK and JNK phosphoacceptor sites in vitro. ERK2, p38 MAPK, and JNK can also phosphorylate PPARγ, while the effect of these MAPKs on PPARβ has not been precisely examined. PKA can phosphorylate all three PPAR isoforms, but the sites have not been accurately mapped. Phosphorylation by the MAPKs or by PKA increased the transcriptional activities of the PPARs, indicating a cooperative rather than antagonistic action of PKA and MAPKs [149].
De novo pyrimidine biosynthetic pathway is controlled by the multifunctional protein CAD (called after the name of its activities: carbamyl-phosphate synthetase, aspartate transcarbamylase, and dihydro-orotase). CAD is activated by ERK1/2-mediated phosphorylation of Thr-456 just prior to the S phase of the cell cycle, when the demand for pyrimidine nucleotides is greatest, and is down-regulated as the cells emerge from S phase by PKA phosphorylation of Ser-1046. Phosphorylation of CAD by PKA antagonizes ERK1/2 phosphorylation, and vice versa. The observation that both MAP kinase and PKA form stable complexes with CAD suggests that the mutual antagonism is the result of steric interference by the bound kinases. The antagonistic properties of ERK1/2 and PKA provides an elegant mechanism to coordinate the cell cycle-dependent regulation of pyrimidine biosynthesis ensuring that signals for up- and down-regulation of the pathway do not conflict [150].
Tubulin polymerization-promoting protein (TPPP/p25) is a brain specific phosphoprotein that displays microtubule bundling activity. ERK2 can phosphorylate TPPP on Ser-18 and Ser-160, while Ser-32 is a phosphoacceptor site for PKA. The phosphorylation of TPPP by ERK2, but not by PKA, perturbed the structural alterations induced by the interaction between TPPP and tubulin [151]. The biological implications of multiple phosphorylation has not been studied.
SCG10, a neuronal-specific stathmin protein with microtubule destabilizing activity, is phosphorylated at Ser-50 and Ser-97 by PKA, and by ERK2 on Ser-62 and Ser-73 in vitro, but all sites have not been confirmed in vivo yet, and the implications of these phosphorylations on the function of SCG10 remain elusive [152].
4 Conclusions
Interconnections between the cAMP/PKA and MAPKs signalling pathways were first described about 15 years ago. These spatial- and temporal-specific integrations allow the cells to tailor cell-specific responses to incoming stimuli. Since then, our knowledge on the molecular mechanisms of cross-talk between these signalling pathways has increased substantially, but still a lot of unanswered questions and conflicting results remain. The use of different methodology (e.g. overexpressing of truncated versions, dominant negative or constitutive active variants of proteins, "specific" inhibitors, RNA interference), different clones of a cell line, or examination of all cell lysates versus subcellular fractions may be responsible for some of these discrepancies. Many challenges lie ahead of us. Novel interactions with less studied MAPKs are likely to be discovered, and these cross-talks may occur in a spatio-temporal manner so that not only time-dependent studies, but also subcellular compartments must be examined. Another puzzling issue is how a cell makes up its mind to what pathways will be engaged and how the correct links are accomplished. While in a football game, a referee assisted by two line men, must ensure that the game proceeds according to the rules, cellular proteins must control the cross-talk events and instruct the "players" how, when and where to interact with each other. Studies with fluorescent-labelled proteins may help us to resolve some of these questions by following specific protein interactions over time in distinct regions of the cell. Understanding how signalling pathways interconnect will not only increase our understanding of cellular functions, it may also aid us in designing more effective therapies for clinical conditions with perturbed signalling transduction pathways. Cancer is a typical signalling transduction disease and several specific inhibitors against protein kinases have entered the clinic or are being tested in clinical trials with promising prospects [153]. Since disturbed action of one protein kinase in a tumour may affect other protein kinases through cross-talk, it may be more advantageous to treat the patient with a cocktail of protein kinase inhibitors.
5 References
Due to space limitations only a selection of the enormous number of relevant articles has been referred. We apologize to those whose work was not mentioned.
T. Hunter, Cell 100 (2000) 113-127.
J.A. Ubersax, J.E. Ferrell Jr, Nat. Rev. Mol. Cell Biol. 8 (2007) 530-541.
D. Houslay, W. Kolch, Mol. Pharmacol. 58 (2000) 659-668.
J.A. Beavo, L.L. Brunton, Nat. Rev. Mol. Cell Biol. 3 (2002) 710-718.
D.M. Cooper, Biochem. Soc. Trans. 33 (2005) 1319-1322.
D.M. Cooper, A.J. Crosswaite, Trends Pharmacol. Sci. 27 (2006) 426-431.
F.D. Smith, L.K. Langeberg, J.D. Scott, Trends Biochem. Sci. 31 (2006) 316-323.
C. Lugnier, Pharmacol. Ther. 109 (2006) 366-398.
Z.G. Goldsmith, D.N. Dhanasekaran, Oncogene 26 (2007) 3122-3142.
M. Aouadi, B. Binetruy, L. Caron, Y. le Marchand-Brustel, F. Bost, Biochimie 88 (2006) 1091-1098.
C. Wellbrock, M. Karasarides, R. Marais, Nat. Rev. Mol. Cell Biol. 5 (2004) 875-885.
J.L. Bos, EMBO J. 17 (1998) 6776-6782 (RAS review!!!!!)
D.T. Leicht, V. Balan, A. Kaplun, V. Singh-Gupta, L. Kaplun, M. Dobson, G. Tzivion.. Biochim. Biophys. Acta 1773 (2007)1196-1212.
A.S. Dhillon, A. von Kriegsheim, J. Grindlay, W. Kolch, Cell Cycle 6(2007) 3-7 =old 53?
S. Ory, M. Zhou, T.P. Conrads, T.D. Veenstra, D.K. Morrison, Current Biol. 13(2003) 1356-1364.
N. Dumaz, R. Marais, J. Biol. Chem. 278 (2003) 29819-29823.
Y.J. Cho, J.Y. Kim, S.W. Jeong, S.B. Lee, O.N. Kim, Leuk. Res. 27 (2003) 51-56.
R.D. York, H. Yao, T. Dillon, C.L. Ellig, S.P. Eckert, E.W. McCleskey, P.J. Stork, Nature 392 (1998) 622-625.
M. Karbowniczek, G.P. Robertson, E.P. Henske, J. Biol. Chem. 281 (2006) 25447-25456.
N.P. Martin, E.J. Whalen, M.A. Zamak, K.L. Pierce, R.J. Lefkowitz, Cell Signal. 16 (2004) 1397-1403.
Y. Daaka, L.M. Luttrell, R.J. Lefkowitz, Nature 390 (1997) 88-91.
R. Pulido, A. Zúñiga, A. Ullrich, EMBO J. 17 (1998) 7337-7350.
M. Gaestel, Nat. Rev. Mol. Cell Biol. 7 (2006) 120-130.
P. Sun, N. Yoshizuka, L. New, B.A. Moser, Y. Li, R. Liao, C. Xie, J. Chen, Q. Deng, M. Yamout, M.Q. Dong, C.G. Frangou, J.R. Yates 3rd, P.E. Wright, J. Han, Cell, 128 (2007) 295-308.
N. Gerits, W. Van Belle, U. Moens, Behav. Brain Funct. 3 (2007) 58.
M.D. Houslay, G. Milligan, Trends Biochem. Sci. 22 (1997) 217-224.
R. Hoffmann, G.S. Baillie, S,J. MacKenzie, S.J. Yarwood, M.D. Houslay, EMBO J. 18 (1999) 893-903.
C. Mehats, C.B. Andersen, M. Filopanti, S.L. Jin, M. Conti, Trends Endocrinol. Metab. 13 (2002) 29-35.
G. Bolger, Cell. Signal. 6 (1994) 851-859.
J.L. Bos, Trends Biochem. Sci. 31 (2006) 680-686. =[13] in old manus!!!!
L.B. Ray, T.W., Sturgill, Proc. Natl. Acad. Sci. USA 84 (1987) 1502-1506.
D.M.F. Cooper, Biochem. J. 375 (2003) 517-529.
Q. Ding, R. Gros, I.D. Gray, R. Tassig, S.S. Ferguson, R.D. Feldman, Mol. Pharmacol. 66 (2004) 921-928.
M.A. Beazely, J.K. Alan, V.J. Watts, Mol. Pharmacol. 67 (2005) 250-259.
M. Hanada, J. Ninomiya-Tsuji, K. Komaki, M. Ohnishi, K. Katsura, R. Kanamura, K. Matsumoto, S. Tamura, J. Biol. Chem. 276(2001) 5753-5759.
M.D. Houslay, Sci. STKE (2006) pe32.
G.S. Baillie, S.J. MacKenzie, I. McPhee, M.D. Houslay, Br. J. Pharmacol., 131 (2000) 811-819.
S.J. MacKenzie, G.S. Baillie, I. McPhee, G.B. Bolgers, M.D. Miles, J. Biol. Chem. 275 (2000) 16609-16617.
M.D. Houslay, M. Sullivan, G. Bolger, Adv. Pharmacol. 44 (1998) 225-342.
M.D. Houslay, Sem. Cell Dev. Biol. 9 (1998) 161-167.
E.V.Hill, C.L. Sheppard, Y.F. Cheung, I. Gall, E. Krause, M.D. Houslay, Cell. Signal. 18 (2006) 2056-2069.
H.T. Zhang, Y. Zhao, Y. Huang, N.R. Dorairaj, L.J. Chandler, J.M. O'Donnell, Neuropsychopharmacology 29 (2004) 1432-1439.
M.T. Koh, T.E. Thiele, I.L. Bernstein, Behav. Neurosci. 116 (2002) 1070-1074.
A.L. Purcell, S.K. Sharma, M.W. Bagnall, M.A. Sutton, T.J. Carew, Neuron 37 (2003) 473-484.
Y. Gao, E. Nikulina, W. Mellado, M.T. Filbin, J. Neurosci. 23 (2003) 11770-11777
F.G. Buchanan, R.N. DuBois, Cell Cycle 5 (2006) 2060-2063.
K. DeFea, Br. J. Pharmacol. (2007) Nov. 26 [Epub ahead of print].
S.M. DeWire, S. Ahn, R.J. Lefkowitz, S.K. Shenoy, Annu. Rev. Physiol. 69 (2007) 483-510.
K.D. Pfleger, M.B. Dalrymple, J.R. Dromey, K.A. Eidne, Biochem. Soc. Trans. 35 (2007) 764-766.
J.D. Violin, R.J. Lefkowitz, Trends Pharmacol. Sci., 28 (2007) 416-422.
L. Ma, G. Pei, J. Cell Sci. 120 (2007) 213-218.
S.J. Perry, G.S. Baillie, T.A. Kohout, I. McPhee, M.M. Magiera, K.L. Ang, W.E. Miller, A.J. McLean, M. Conti, M.D. Houslay, R.J. Lefkowitz, Science 298 (2002) 834-836.
G.B. Bolger, A. McCahill, E. Huston, Y.F. Cheung, T. McSorley, G.S. Baillie, M.D. Houslay, J. Biol. Chem. 278 (2003) 49230-49238.
G.S. Baillie, A. Sood, I. McPhee, I. Gall, S.J. Perry, R.J. Lefkowitz, M.D. Miles, Proc. Natl. Acad. Sci. USA 100 (2003) 940-945.
M.J. Lynch, G.S. Baillie, A. Mohamed, X. Li, C. Maisonneuve, E. Klussmann, G. van Heeke, M.D. Houslay, J. Biol. Chem. 280v(2005) 33178-33189.
D. Willoughby, G.S. Baillie, M.J. Lynch, A. Ciruela, M.D. Houslay, D. M.F. Cooper, J. Biol. Chem. 282 (2007) 34235-34249.
G.B. Bolger, G.S. Baillie, X. Li, M.J. Lynch, P. Herzyk, A. Mohamed, L.H. Mitchell, A. McCahill, C. Hundsrucker, E. Klussmann, D.R. Adams, M.D. Houslay, Biochem. J. 398 (2006) 23-36.
K.J. Smith, G.S. Baillie, E.I. Hyde, X. Li, T.M. Houslay, A. McCahill, A.J. Dunlop, G.B. Bolger, E. Klussmann, D.R. Adams, M.D. Houslay, Cell. Signal. 19 (2007) 2612-2624.
[1] B.S. Skålhegg, K. Taskén, Front. Biosci. 5 (2000) 678-693.
[2] K. Taskén, E.M. Aandahl, Physiol. Rev. 84 (2004) 137-167.
[3] S.S. Taylor, C. Kim, D. Vigil, N.M. Haste, J. Yang, J. Wu, G.S. Anand, Biochim. Biophys. Acta 1754 (2005) 25-37.
[4] R. Das, V. Esposito, M. Abu-Abed, G.S. Anand, S.S. Taylor, G. Melacini, Proc. Natl. Acad. Sci. USA 104 (2007) 93-98.
[5] M.D. Houslay, C. Milligan. Trends Biochem. Sci. 22 (1997) 217-224.
[6] M.D. Houslay and D.R. Adams. Biochem. J. 370 (2003) 1-18.
[9] N. Pham, I. Cheglakov, C.A. Koch, C.L. de Hoog, M.F. Moran, D. Rotin, Curr. Biol., 10 (2000) 555-558.
[10] Y. Pak, N. Pham, D. Rotin, Mol. Cell. Biol. 22 (2002) 7942-7952.
[11] U.B. Kaupp, R. Seifert, Physiol. Rev. 82 (2002) 769-824.
[12] S. Dremier, R. Koopperud, S.O. Døskeland, J.E. Dumont, C. Maenhaut, FEBS Lett. 546 2003) 103-107.
[13] J.L. Bos, Trends Biochem. Sci. 31 (2006) 680-688.
[14] G.L. Johnson, R. Lapadat, Science 298 (2002) 1911-1912.
[15] P.P. Roux, J. Blenis, Microbiol. Mol. Biol. Rev. 68 (2004) 320-344.
[16] M. Imajo, Y. Tsuchiya, E. Nishida, IUBMB Life 58 (2006) 312-317.
[17] A. Cuenda, S. Rousseau, Biochim. Biophys. Acta 1773 (2007) 1358-1375.
[18] B. Cuevas, A. Abell, G. Johnson, Oncogene 26 (2007) 3159-3171.
[19] M. Raman, W. Chen, M.H. Cobb, Oncogene 26 (2007) 3100-3112.
[20] C.R. Weston, R.J. Davis, Curr. Opin. Cell. Biol. 19 (2007) 142-149.
[21] Y. Zhang, C. Dong, Cell. Mol. Life Sci. 64 (2007) 2771-2789.
[22] J.L. Bos, H. Rehmann, A. Wittinghofer, Cell 129 (2007) 865-877. Erratum in Cell 130 (2007) 385.
[23] N. Gerits, S. Kostenko, U. Moens, Trans. Res. 16 (2007) 281-314.
[24] D.M. Vogt Weisenhorn, L.J. Roback, J.H. Kwon, B.H. Wainer. Exp. Neurol. 169 (2001) 44-55.
[25] B.M.T. Burgering, G.J. Pronk, P.C. van Weeren, P. Chardin, and J.L. Bos, EMBO J. 12 (1993) 4211-4220.
[26] S. J. Cook, F. McCormick, Science 262 (1993) 1069-1072.
[27] L.M. Graves, K.E. Bornfeldt, E.W. Raines, B.C. Potts, S.G. MacDonald, R. Ross, E.G. Krebs, Proc. Natl. Acad. Sci. USA 90 (1993) 10300-10304.
[28] B.R. Sevetson, X. Kong, J.C. Lawrence Jr, Proc. Natl. Acad. Sci. USA 90 (1993) 10305-10309.
[29] J. Wu, P. Dent, T. Jelinek, A. Wolfman, M.J. Weber, T.W. Sturgill, Science 262 (1993) 1065-1069.
[30] M. Frödin, P. Peraldi, E. Van Obberghen, J. Biol. Chem. 269 (1994) 6207-6214.
[31] S.W. Young, M. Dickens, J.M. Tavaré, FEBS Lett. 338 (1994) 212-216.
[32] F. Lamy, F. Wilkin, M. Baptist, J. Posada, R.R. Roger, J.E. Dumont, J. Biol. Chem. 268 (1993) 8398-8401.
[33] D.J. Creedon, E.M. Johnson, J.C. Lawrence Jr, J. Biol. Chem. 271 (1996) 20713-20718.
[34] F.R. McKenzie, J. Pouyssegur, J. Biol. Chem. 271 (1996) 13476-13483.
[35] P. Erhardt, J. Troppmair, U.R. Rapp, G.M. Cooper, Mol. Cell. Biol. 15 (1995) 5524-5230.
[36] N. Mitin, K.L. Rossman, C.J. Der, Curr. Biol. 15 (2005) R563-574.
[37] K. Rajalingam, R. Schreck, U.R. Rapp, S. Albert, Biochim. Biophys. Acta 1773 (2007) 1177-1195.
[38] A. Ambrosini, S. Tininini, A. Barassi, G. Racagni, E. Sturani, R. Zippel, Brain Res. Mol. Brain Res. 75 (2000) 54-60.
[39] R. Buscà, P. Abbe, F. Mantoux, E. Aberdam, C. Peyssonnaux, A. Eychène, J.P. Ortonne, R. Ballotti, EMBO J. 19 (2000) 2900-2910.
[40] O.M. Tsygankova, E. Kupperman, W. Wen, J.L. Meinkoth, Oncogene 19 (2000) 3609-3615.
[41] N. Iida, K. Namikawa, H. Kiyama, H. Ueno, S. Nakamura, S. Hattori, J. Neurosci. 21 (2001) 6459-6466.
[42] E.M. Amsen, N. Pham, Y. Pak, D. Rotin, J. Biol. Chem. 281 (2006) 121-128.
[43] J.H. Norum, T. Methi, R.R. Mattingly, F.O. Levy. FEBS J 272 (2005) 2304-2316.
[44] L.A. Quilliam, J.F. Rebhun, A.F. Castro, Prog. Nucleic Acid Res. Mol. Biol. 71 (2002) 391-444.
[45] P. Peraldi, M. Frödin, J.V. Barnier, V. Calleja, J.C. Scimeca, C. Filloux, G. Calothy, E. Van Obberghen. FEBS Lett. 357 (1995) 290-296.
[46] R.A. McPherson, A. Harding, S. Roy, A. Lane, J.F. Hancock, Oncogene 18 (1999) 3862-3869.
[47] M.D. Houslay and W. Kolch, Mol. Pharmacol. 58 (2000) 659-668
[48] A.S. Dhillon, C. Pollock, H. Steen, P.E. Shaw, H. Mischak, W. Kolch, Mol. Cell. Biol. 22 (2002) 3237-3246.
[49] N. Dumaz, Y. Light, R. Marais, Mol. Cell. Biol. 22 (2002) 3717-3728.
[50] H. Chong, H.G. Vikis, K.L. Guan. Cell. Signal. 15 (2003) 463-469.
[51] M. Baccarini, FEBS Lett. 579 (2005) 3271-3277.
[52] N. Dumaz, R. Marais, FEBS J. 272 (2005) 3491-3504.
[53] A.S. Dhillon, A. von Kriegsheim,J. Grindlay, W. Kolch, Cell Cycle 6 (2007) 3-7.
[54] M.F. Sidovar, P. Kozlowski, J.W. Lee, M.A: Collins, Y. he, L.M. Graves, J. Biol. Chem. 275 (2000) 28688-28694.
[55] M.L. Edin, R.L. Juliano, Mol. Cell. Biol. 25 (2005) 4466-4475.
[56] H. Mischak, T. Steitz, P. Janosch, M. Eulitz, H. Steen, M. Schellerer, A. Philipp, W. Kolch, Mol. Cell. Biol. 16 (1996) 5409-5418.
[57] J.H. Ahn, T. McAvoy, S.V. Rakhilin, A. Nishi, P. Greengard, A.C. Nairn, Proc. Natl. Acad. Sci. USA. 104 (2007) 2979-2984.
[58] T. Brummer, P. Martin, S. Herzog, Y. Misawa, R.J. Daly, M. Reth, Oncogene 25 (2006) 6262-6276.
[59] Qiu, W., S. Zhuang, F.C. von Lintig, G.R. Boss, R.B. Pilz, J. Biol. Chem. 275 (2000) 31921-31929.
[60] M.C. MacNicol, A. M. MacNicol, J. Biol. Chem. 274 (1999) 13193-13917.
[61] B.H. Zhang, K.L. Guan, EMBO J. 19 (2000) 5429-5439.
[62] S.J. Cook, B. Rubinfeld, I. Albert, F. McCormick, EMBO J. 12 (2003) 3475-3485.
[63] J.M. Schmitt, P.J.S. Stork, Mol Cell Biol. 21(2001) 3671-3683.
[64] I. Lerosey, V. Pizon, A. Tavitian, J. de Gunzburg, Biochem. Biophys. Res. Commun. 175 (1991) 430-436.
[65] L.A. Quilliam, H. Mueller, B.P. Bohl, V. Prossnitz, L.A. Sklar, C.J. Der, G.M. Bokoch, J. Immunol. 147 (1991) 1628-1635. Erratum in: J. Immunol. 147 (1991) 3660.
[66] P. Polakis, B. Rubinfeld, F. McCormick, J. Biol. Chem. 267 (1992) 10780-10785.
[67] M.R. Vossler, H. Ya, R.D. York, M.G. Pan, C.S. Rim, P.J.S. Stork, Cell 89 (1997) 73-82.
[68] D.L. Altschuler, S.N. Peterson, M.C. Ostrowski, E.G. Lapetina, J Biol. Chem. 270 (1995) 10373-10376.
[69] M. Klinger, O. Kudlacek, M.G. Seidel, M. Freissmuth, V. Sexl, J. Biol. Chem. 277 (2002) 32490-32497.
[70] J.M. Schmitt, P.J.S. Stork, Mol. Cell 9 (2002) 85-94.
[71] Y. Obara, K. Labudda, T.J. Dillon, P.J. Stork, J. Cell. Sci. 117 (2004) 6085-6094.
[72] H. Yao, R.D. York, A. Misra-Press, D.W. Carr, P.J.S. Stork, J. Biol. Chem. 273 (1998) 8240-8247.
[73] J.M. Enserink, A.E. Christensen, J. de Rooij, M. van Triest, F. Schwede, H.G. Genieser, S.O. Døskeland, J.L. Blank, J.L. Bos, Nat. Cell Biol. 4 (2002) 901-906.
[74] M.P. Delghandi, M. Johannessen, U. Moens, Cell. Signal. 17 (2005) 1343-1351.
[75] Z. Wang, T.J. Dillon, V. Pokala, S. Mishra, K. Labudda, B. Hunter, P.J.S. Stork, Mol. Cell. Biol. 26 (2006) 2130-2145.
[76] Y. Li, S. Asuri, J.F. Rebhun, A.F. Castro, N.C. Paranavitana, L.A. Quilliam, J. Biol. Chem. 281 (2006) 2506-2514.
[77] Y. Obara, A.M. Horgan, P.J.S. Stork, J. Neurochem. 101 (2007) 470-482.
[78] A. Morozov, I. A. Muzzio, R. Bourtchouladze, N. Van Strien, K. lapidus, D. Yin, D.G. Winder, J.P. Adams, J.D. Sweatt, E.R. Kandel, Neuron 39 (2003) 309-325.
[79] L. Wang, F. Liu, M.L. Adamo, J. Biol. Chem. 276 (2001) 37242-37249.
[80] P.J.S. Stork, J.M. Schmitt, Trends Cell Biol. 12 (2002) 258-266.
[81] W.M. Yee, P.F. Worley, Mol. Cell. Biol. 17 (1997) 921-933.
[82] R.J. Lefkowitz, K.L. Pierce, L.M. Luttrell, Mol. Pharmacol. 62 (2002) 971-874.
[83] M. Keiper, M.B. Stope, D. Szatkowski, A. Böhm, K. Tysack, F. vom Dorp, O. Saur, P.A.O. Weernink, S. Evellin, K.H. Jakobs, M. Schmidt, J. Biol. Chem. 279 (2004) 46497-46508.
[84] G. Tortora, V. Damanio, G. Bianco, G. Baldasarre, A.R. Bianco, L. Lanfrancone, P.G. Pelicci, F. Ciardiello, Oncogene 14 (1997) 923-928.
[85] H. Sun, Z. Chen, H. Poppleton, K. Scholich, J. Mullenix, G.J. Weipz, D.L. Fulgham, P.J. Bertics, T.B. Patel, J. Biol. Chem. 272 (1997) 5413-5420.
[86] M. Saxena, S. Williams, K. Taskén, T. Mustelin, Nat. Cell Biol. 1 (1999) 305-311.
[87] M. Hunzicker-Dunn, E.T. Maizels, Cell. Signal. 18 (2006) 1351-1359.
[88] L.M. Graves, K.E. Bornfeldt, J.S. Sidhu, G.M. Argast, E.W. Raines, R. Ross, C.C. Leslie, E.G. Krebs. J. Biol. Chem. 271 (1996) 505-511.
[89] A. Rahman, K.N. Anwar, M. Minhajuddin, K.M. Bijli, K. Javaid, A.L. True, A.B. Malik, Am. J. Physiol. Lung Cell. Mol. Phys. 287 (2004) L1017-1029.
[90] J. Ster, F. De Bock, N.C. Guérineau, A. Janossy, S. Barrère-Lemaire, J.L. Bos, J. Bockaert, L. Fagni, Proc. Natl. Acad. Sci. USA 104 (2007) 2519-2524.
[91] J. Jensen, Br. J. Pharmacol. 151 (2007) 423-425.
[92] Y. Kanda, Y. Watanabe, Br. J. Pharmacol. 151 (2007) 476-482.
[93] C.C. Huang, J.L. You, M.Y. Wu, K.S. Hsu, J. Biol. Chem. 279 (2004) 12286-1292.
[94] Y. Sawada, K. Nakamura, D. Doi, K. Takeda, K. Tobiume, M. Saitoh, I. Komuro, K. De Vos, M. Sheetz, H. Ichijo, J. Cell Sci. 114 (2001) 1221-1227.
[95] R. Pulido, A. Zúñiga, A. Ullrich, EMBO J. 17 (1998) 7337-7350.
[96] M. Oh-hora, M. Ogata, Y. Mori, M. Adachi, K. Imai, A. Kosugi, T. Hamaoka, J. Immunol. 163 (1999) 163 1282-1288.
[97] K. Nika, H. Hyunh, S. Williams, S. Paul, N. Bottini, K. Taskén, P.J. Lombroso, T. Mustelin, Biochem. J. 378 (2004) 335-342.
[98] C. Blanco-Aparicio, J. Torres, R. Pulido, J. Cell Biol. 6 (1999) 1129-1135.
[99] J.J. Zhu, Y. Qin, M. Zhao, L. Van Aelst, R. Malinow, Cell 110 (2002) 443-455.
[100] J. Robidoux, W. Cao, H. Quan, K.W. Daniel, F. Moukdar, X. Bai, L.M. Floering, S. Collins, Mol. Cell. Biol. 25 (2005) 5466-5479.
[101] P.C. Cheung, A.R. Nebreda, P. Cohen, Biochem. J. 378 (2004) 27-34.
[102] R. Lim, A. Zaheer, Khosravi, H., J.H. Freeman Jr, H.E. Halverson, J.A. Wemmie, B. Yang, Brain Res 1024 (2004) 225-232.
[103] R. Lim, A. Zaheer, J. Biol. Chem. 271 (1996) 22953-22956.
[104] A. Zaheer, R. Lim, Biochem. Biophys. Res. Commun. 250 (1998) 278-282.
[105] A. Zaheer, M.A. Yorek, R. Lim, Neurochem. Res. 26 (2001) 1293-1299.
[106] R.A. Cardone, A. Bagorda, A. Bellizzi, G. Busco, L. Guerra, A. Paradiso, V. Casavola, M. Zaccolo, S.J. Reshkin, Mol. Biol. Cell 16 (2005) 3117-3127.
[107] R.A. Cardone, A. Bellizzi, G. Busco, E.J. Weinmann, M.E. Dell'Aquil, V. Casavola, A. Azzariti, A. Mangia, A. Paradiso, S.J. Reshkin, Mol. Biol. Cell 18 (2007) 1768-1780.
[108] G.X. Shi, H. Rehmann, D.A. Andres, Mol. Cell. Biol. 26 (2006) 9136-9147.
[109] J.L. Rudolph, G.X. Shi, E. Erdogan, A.P. Fields, D.A. Andres, Biochim. Biophys. Acta 1773 (2007) 1793-1800.
[110] Y.P. Hsueh, M.Z. Lai, J. Biol. Chem. 270 (1995) 18094-18098.
[111] A. Tamir, Y. Granot, N. Isakov, J. Immunol. 157 (1996) 1514-1522.
[112] S.A. Black Jr., A.H. Palamakumbura, M. Stan, P.C. Trackman, J. Biol. Chem. 282 (2007) 15416-15429.
[113] H.J. Chae, S.W. Chae, H.R. Kim, Immunopharmacol. Immunotoxicol. 26 (2004) 249-263.
[114] A.L. Miller, A.S. Garza, B.H. Johnson, E. B. Thompson, Cancer Cell Int. 7 (2007) 3.
[115] C. Burgun, L. Esteve, N. Humblot, D. Aunis, J. Zwiller, FEBS Lett. 484 (2000) 189-193.
[116] A. Sommer, H. Burkhardt, S.M. Keyse, B. Lüscher, FEBS Lett. 474 (2000) 146-150.
[117] D.M. Owens, S.M. Keyse, Oncogene 26 (2007) 3203-3213.
[118] M. Delgado, Biochem. Biophys. Res. Comm. 293 (2002) 771-776.
[119] E.D. Gallagher, S. Xu, C. Moomaw, C.A. Slaughter, M.H. Cobb, J. Biol. Chem. 277 (2002) 45785-45792.
[120] H. Xu, S. Washington, M.F. Verderame, A. Manni, Breast Cancer Res. Treat. (2007) [Epub ahead of print].
[121] D. Chen, S. Reierstad, Z. Lin, M. Lu, C. Brooks, N. Li, J. Innes, S.E. Bulun, Cancer Res. 67 (2007) 8914-8922.
[122] K. Kurokawa, K. Kawai, H. Hashimoto, Y. Ito, M. Takahashi, J. Intern. Med. 253 (2003) 527-533.
[123] N. Asai, T. Fukuda, Z. Wu, A. Enomoto, V. Pachnis, M. Takahashi, F. Costantini, Development 133 (2006) 4507-4516.
[124] D. Hochbaum, T. Tanos, F. Ribeiro-Neto, D. Altschuler, O.A. Coso, J. Biol. Chem. 278 (2003) 33738-33746.
[125] N. Mochizuki, Y. Ohba, S. Kobayashi, N. Otsuka, A. M. Graybiel, S. Tanaka, M. Matsuda, J. Biol. Chem. 275 (2000) 12667-12671.
[126] S. Fuld, G. Borland, S.J. Yarwood, Exp. Cell. Res. 309 (2005) 161-173.
[127] K.L. Dodge-Kafka, J. Soughayer, G.C. Pare, J.J.C. Michel, L.K. Langeberg, M.S. Kapiloff, J.D. Scott, Nature 437 (2005) 574-578.
[128] K.L. Dodge-Kafka, M.S. Kapiloff, Eur. J. Cell Biol. 85 (2006) 593-602.
[129] G.W. Pearson, M.H. Cobb, J. Biol. Chem. 277 (2002) 48094-48098.
[130] G.W. Pearson, S. Earnest, M.H. Cobb, Mol. Cell. Biol. 26 (2006) 3039-3047.
[131] D.Chaturvedi, H.M. Poppleton, T. Springfield, A. Barbier, T.B. Patel, Mol. Cell. Biol. 26(2006) 4586-4600.
[132] P.P. Roux, B.A. Ballif, R. Anjum, S.P. Gygi, J. Blenis. Proc. Natl. Acad. Sci. USA 101 (2004) 13489-13494.
[133] D.J. Kwiatkowski, B.D. Manning, Hum. Mol. Gen. 14 (2005) R1-R8.
[134] R. Wienecke, A. Guha, J.C. Maize, R.L. Heideman, J.E. DeClue, D.H. Gutmann, Ann. Neurol. 42 (1997) 230-235.
[135] J.A. Rudolph, J. Pratt, R. Murya, K.A. Steinbrecher, M.B. Cohen, Cell. Signal. 19 (2007) 1221-1228.
[136] O.M. Seternes, B. Johansen, B. Hegge, M. Johannessen, S.M. Keyse, U. Moens, Mol. Cell. Biol. 22 (2002) 6931-6945.
[137] N. Gerits, T. Mikalsen, S. Kostenko, A. Shiryaev, M. Johannessen, U. Moens, J. Biol. Chem. 282 (2007) 37232-37243.
[138] P. Sun, N. Yoshizuka, L. New, B.A. Moser, Y. Li, R. Liao, C. Xie, J. Chen, Q. Deng, M. Yamout, M.Q. Dong, C.G. Frangou, J.R. Yates 3rd, P.E. Wright, J. Han, Cell 128 (2007) 295-308.
[139] H. Liu, D. Palmer, S.L. Jimmo, D.G. Tilley, H.A: Dunkerley, S.C. Pang, D.H. Maurice, J. Biol. Chem. 275 (2000) 26615-26624.
[140] M.D. Houslay, G.S. Baillie, Biochem. Soc. Trans. 31 (2003) 1186-1190.
[141] M. Johannessen and U. Moens, Front Biosci. 12 (2007)12:1814-1832.
[142] C.B. Sindreu, Z.S. Scheiner, D.R. Storm, Neuron 53 (2007) 79-89.
[143] S.R. Datta, A. Brunet, M.E. Greenberg, Genes Dev 13 (1999) 2905-2927.
[144] X. Pi, C. Yan, B.C. Berk, Circ. Res. 94 (2004) 362-369.
[145] J. Wu, R. Janknecht, J. Biol. Chem. 277 (2002) 42669-42679.
[146] K. Toska, R. Kleppe, C.G.. Armstrong, N.A. Morrice, P. Cohen, J. Haavik, J. Neurochem 83 (2002) 775-783.
[147] J. Wu, D. Filer, A.J. Friedhoff, M. Goldstein, J. Biol. Chem. 267 (1992) 23754-23758.
[148] K. Toska , R. Kleppe, P. Cohen, J. Haavik, Ann N Y Acad Sci. 971 (2002b) 66-68.
[149] K.A. Burns, J.P. Vanden Heuvel, Biochim. Biophys. Acta 1771 (2007) 952-960.
[150] F.D. Sigoillot, D.H. Kotsis, E.M. Kotsis, E.M. Masko, M. Bame, D.R. Evans, H.I. Evans, Front. Biosci. 12 (2007) 3892-3898.
[151] E. Hlavanda, E. Klement, E. Kókai, J. Kovács, O. Vincze, N. Tökési, F. Orosz, K.F. Medzihradszky, V. Dombrási, J. Ovádi, J. Biol. Chem. 282 (2007) 29531-29539.
[152] H. Morii, T. Yamada, I. Nakano, J.M. Coulson, N. Mori, Neurosci. Lett. 396 (2006) 241-246.
[153] T. Mikalsen, N. Gerits, U. Moens, Biotechnol. Annu. Rev. 12 (2006) 153-223.