|
*Corresponding author: Nancy Gerits; Department of Microbiology and Virology, Institute of Medical Biology, University of Tromsø, N-9037 Tromsø, Norway. Tel.: +47-776 44733; fax: +47-776 45350; e-mail: nancy@sigtrans.org
Keywords : mice, knock-in, knock-out, MAP kinase, pathogens, anti-MAP kinase therapies, genetic background, phenotype, inflammatory diseases, cancer
AbstractMulticellular organisms achieve intercellular communication by means of signalling molecules whose effect on the target cell is mediated by signal transduction pathways. Such pathways relay, amplify and integrate signals to elicit appropriate biological responses. Protein kinases form crucial intermediate components of numerous signalling pathways. One family of protein kinases, the mitogen-activated protein kinases (MAP kinases) are kinases involved in signalling pathways that respond primarily to mitogens and stress stimuli. In vitro studies revealed that the MAP kinases are implicated in several cellular processes, including cell division, differentiation, cell survival/apoptosis, gene expression, motility and metabolism. As such, dysfunction of specific MAP kinases is associated with diseases such as cancer and immunological disorders. However, the genuine in vivo functions of many MAP kinases remain elusive. Genetically modified mouse models deficient in a specific MAP kinase or expressing a constitutive active or a dominant negative variant of a particular MAP kinase offer valuable tools for elucidating the biological role of these protein kinases. In this review, we focus on the current status of MAP kinase knock-in and knock-out mouse models and their phenotypes. Moreover, examples of the application of MAP kinase transgenic mice for studying human clinical conditions, for validating therapeutic properties of specific MAP kinase inhibitors, and for investigating the role of MAP kinase in pathogen-host interactions will be discussed. Contents 1 Introduction
7 Applications
of MAP kinase TG mice in medical research 2 An overview of the MAP kinase signalling pathways
7.1 MAP kinase mouse models in host-pathogen infection research 7.2 MAP kinase mouse models to develop anti-MAP kinase therapy 8 Conclusion |
Protein kinases and phosphatases regulate the activity of proteins respectively by reversible phosphorylation and dephosphorylation. The finding that approximately 30% of all the proteins in cells are phosphoproteins and that more than 2% of the human genes encode protein kinases (~520 genes) and protein phosphatases (~150 genes) underscores the biological significance of this transient protein modification. Many protein kinases and phosphatases participate in signalling pathways that mediate communication between cells and regulate cellular processes in response to specific signals. The family of mitogen-activated protein kinases (MAP kinases1) consists of a large family of protein kinases implicated in cellular processes such as gene regulation, metabolic reactions, cell proliferation, cell differentiation, cell mobility and cell survival or cell death [Roux and Blenis, 2004]. By consequence, perturbed action of the MAP kinases contributes to cancer, diabetes and inflammatory diseases. Additionally, infection by pathogens can also target and impair the activity of these proteins [Mourin and Huot, 2004, Münter et al., 2006].
Biochemical studies and experiments with ectopic expression of dominant negative and constitutive active mutants, RNA interference, and specific protein kinase inhibitors in cell cultures still contribute enormously to unravelling the constitution of the MAP kinase signalling pathways. However, studies with transgenic mice and naturally occurring genetic diseases offer help to understand the biological role of these kinases in vivo. This review focuses on the phenotypes obtained by construction of transgenic mouse models of the different MAP kinases. On the other hand, the use of cell lines derived from such mice and the studies on establishing the position of a specific MAP kinase in signalling pathways fall beyond the scope of this review. Transgenic mice not only provide essential information on the biological function of the specific MAP kinase, they are also valuable models to identify candidate genes implicated in human diseases and to unravel the molecular mechanisms involved in pathogenic processes. In addition, these mice can be used to test specific inhibitors against MAP kinases designed for therapeutic purposes. Finally, as many pathogens engage MAP kinase pathways for their successful infection, MAP kinase transgenic mice can be used to increase our knowledge on the molecular basis of pathogen-host infections and to develop strategies to prevent or abort such infections.
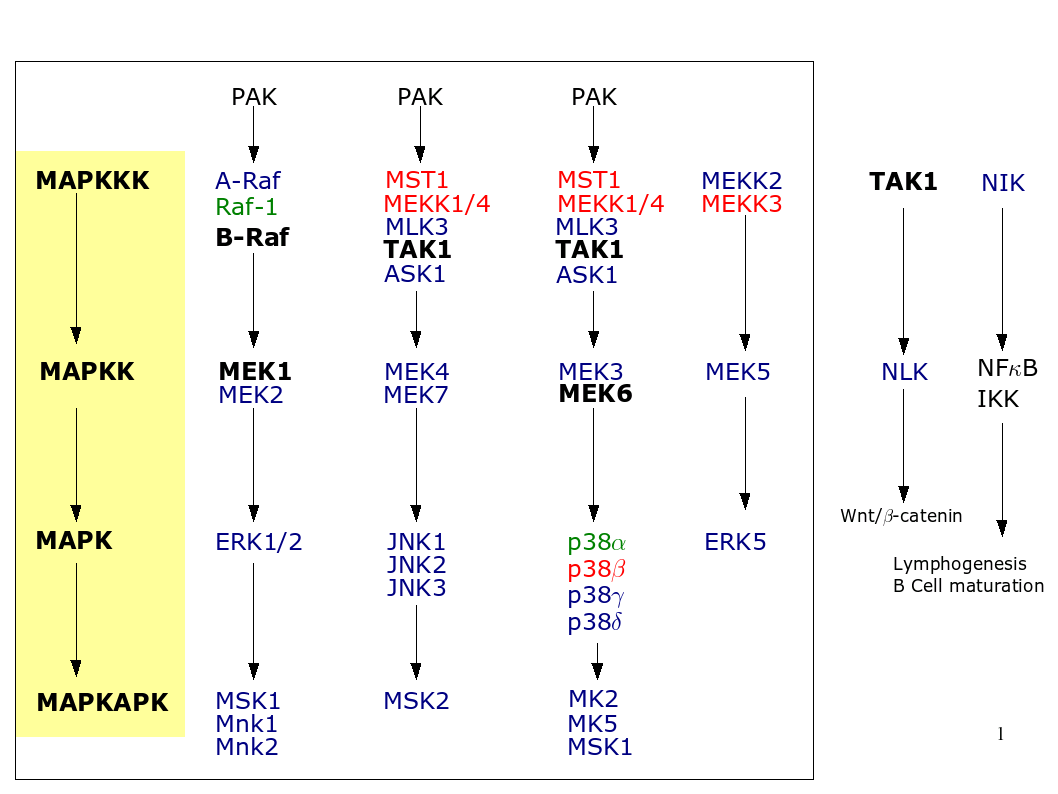
A typical MAP kinase module encompasses a cascade of three kinases where MAP kinase kinase kinase (MAPKKK) phosphorylates and activates a MAP kinase kinase (MAPKK), which in turn phosphorylates a MAP kinase. MAP kinases can either phosphorylate non-kinase proteins such as transcription factors or yet other kinases referred to as MAP kinase-activating protein kinases or MK. Figure 1, summarizes the different MAP kinase pathways and shows for which MAP kinase there exists a mouse model. We briefly discuss each of the pathways in this section.
|
Additionally, Table 1 summarizes the phenotypical changes observed in MAP kinase knock-in (KI) and knock-out (KO) mice.
Also known as the classical MAP kinase signalling pathway, the ERK pathway consists of the MAPKKKs A-Raf, B-Raf, and c-Raf-1, the MAPKKs MEK1 and MEK2, the MAPKs ERK1 and ERK2, and the MAPKAPKs MNK1, MNK2, MSK1, MSK2, RSK1/2/3, p70S6K and p70S5K. This pathway plays important roles in cellular processes like proliferation, differentiation and survival. Cytokines, growth factors, serum, certain stresses, ligands for G protein-coupled receptors and microtubule disorganization can all activate A-Raf, B-Raf and c-Raf-1. Those kinases then differentially regulate the activity of their downstream effectors MEK1 and MEK2 by phosphorylation. These highly homologous isoforms phosphorylate the Thr-Glu-Tyr motif in the activation loop of the ERKs. ERK1 and ERK2, respectively 44 and 42 kDa in size, share 90% sequence identity and target numerous proteins, like transcription factors (e.g. STAT, Ets, Elk-1, oestrogen receptor) and cytoplasmic proteins (e.g. phospholipase A2) [Roux and Blenis, 2004].
The JNKs were originally discovered as kinases that could phosphorylate the NH2-terminal part of the c-Jun transcription factor. Stimuli that activate Rho family receptors, tyrosine kinase receptors or cytokine receptors, transmit the signal to the upstream MAPKKKs like MLKs, ALK, TAK, and TLP. Those MAPKKKs then phosphorylate MAPKKs MKK4 and MKK7, which subsequently phosphorylate and activate JNK1, JNK2 and JNK3. These MAP kinases occur in different splice forms as 46kDa or 55kDa proteins, depending on the absence or presence of a C-terminal tail whose function remains unknown. JNK1 and JNK2 are ubiquitously expressed, whereas JNK3’s expression is mainly limited to the brain, heart and testis. The physiological significance of the JNK pathway lays in apoptotic and survival pathways, embryogenic morphogenesis, tumour biology and immunological diseases, however the mechanisms underlaying its involvement remain to be established [Davis, 2000, Imajo et al., 2006].
The discovery of the p38 pathway started when stimulation of macrophages with LPS led to the tyrosine phosphorylation of a protein of 38kDa, p38a, and its subsequent characterization. Similar to the JNK pathway, the p38 pathway becomes activated by environmental stress and shares approximately the same upstream MAPKKK activators. Again the difference lays at the level of the MAPKKs (MKK3 and MKK6) and their downstream effectors: MKK6 activates all p38 MAP kinases, whereas MKK3 only activates p38a and p38b [Roux and Blenis, 2004, Imajo et al., 2006].
The p38 MAPKs consist of p38a, p38b, p38g and p38d. p38a and p38b share 74% amino acid sequence homology and their wide expression pattern. p38g and p38d share respectively 63% and 61% of their amino acid sequence with p38a. Both kinases have a more limited expression pattern: The former being mainly expressed in skeletal muscle, whereas the latter can be found predominantly in testis, pancreas, small intestine, and CD4+ T-cells. Aside from their high sequence identity, they also share wide substrate similarity, although the activity towards these same substrates can differ substantially. In the end, each kinase has some specific substrates as well. The very diverse p38 pathway plays roles in regulating the immune systems response, mammalian preimplantation development, cell survival and death, differentiation and growth [Imajo et al., 2006, Ashwell, 2006, Wada and Penninger, 2004].
The 98kDa protein, Big MAP kinase1 (BMK1 or ERK5), earned its name for its big size, compared to the related ERK1 and ERK2. The protein exists in three splice forms, whose biological significance remains to be fully explored. Two of the isoforms may act in a dominant-negative fashion on ERK5, thereby allowing additional fine tuning of the MEK5/ERK5 pathway. ERK5 is ubiquitously expressed, except for the liver. The kinase shares 66%-67% sequence similarity with the kinase domains of ERK1/2 and oxidative stress, as well as mitogenic stimuli can activate ERK5. Both MEKK2 and MEKK3 seem to activate the unique upstream activator of ERK5, namely MEK5, via phosphorylation. MEK5 on its turn then phosphorylates and activates ERK5. Downstream substrates of ERK5 include transcription factors and the serum- and glucocorticoid-inducible kinase (reviewed in Wang and Tournier, 2006, Bogoyevitch and Court, 2004).
Little is known about the signalling cascades involving the ERKs 3, 4, 6, 7 and 8. ERK3 (MAP kinase 6) interacts with and phosphorylates MAPKAPK5 in vitro and in vivo and functions as a substrate for this kinase as well. MEKs do not seem to phosphorylate ERK3, so that the upstream activators of ERK3 remain elusive [Schumacher et al., 2004, Seternes et al., 2004]. The poorly characterized 45 kDa ERK4 protein has been suggested to be the splice form ERK1b that contains a 26 amino acid insertion [Bogoyevitch and Court, 2004]. Its upstream activators and target substrates remain unidentified, although reports indicated a direct interaction between ERK4 and MAPKAPK5 [Schumacher et al., 2004].
The amino acid sequence of the kinase domain of ERK7, a 61 kDa protein, shares approximately 40% homology with ERK1 and ERK2, but neither MEK1 nor MEK2 phosphorylate ERK7. Which kinase does remains currently unknown, instead ERK7 seems to be constitutively active due to autophosphorylation. ERK7 phosphorylates the transcription factors c-Fos and c-Myc, and interacts directly with but doesn’t phosphorylate the intracellular chloride channel CLIC3. Some evidence indicates that ERK7 also phosphorylates the oestrogen receptor-a [Bogoyevitch and Court, 2004, Henrich et al., 2003].
With its 69% overall amino acid identity (82% in the kinase domain), ERK8 closely resembles ERK7. Thus far, only serum seems to activate ERK8, in a Src-dependent way (reviewed in Bogoyevitch and Court 2004). Recently, it was shown that RET/PTC3, a constitutively active form of RET, activated ERK8 and that ERK8 directly interacts with and regulates the activity of Hic-5/ARA55, a LIM protein that functions as a cofactor for nuclear receptors [Iavarone et al., 2006, Saelzler et al., 2006]
TNFa, Fas ligand and stress activate ASK1, which then phosphorylates MKK3/MKK6 and MKK4/MKK7. This results ultimately in the activation of the p38 and JNK MAP kinase pathway, respectively (see Figure 1; Matsuzawa et al., 2002, Nishitoh et al., 2002).
Homozygous dnASK1Dexon1 mice were born at the expected Mendelian frequency, appeared indistinguishable from their WT littermates and revealed no developmental abnormalities as confirmed by histological analysis [Tobiume et al., 2001]. These mice also showed normal basal body mass, blood pressure, heart rate and basal cardiac function and dimensions. However, angiotensin II-induced cardiac hypertrophy, cardiomyocyte apoptosis, myocardial interstitial and coronary arterial thickening, perivascular fibrosis, and expression of cardiac genes were significantly attenuated in ASK1-∕- mice compared with ASK+∕+ mice [Izumiya et al., 2003].
Moreover, ASK1-∕- mice showed decreased ventricular remodelling after myocardial injury induced by coronary artery ligation or TAC as revealed by their increased lung weight and lung-to-body weight ratios four weeks after treatment [Yamaguchi et al., 2003]. Additionally, the ASK1-∕- mice suffered from reduced ischemia-induced angiogenesis. Comparative studies between WTs and ASK1-∕- mice revealed that ASK1 plays a role in the intimal thickening after vascular injury as well [Izumi et al., 2003, Izumi et al., 2005]. These observations suggest that ASK1 plays an important role in regulating LV remodelling by promoting apoptosis. Hence, it may form a target for novel drugs to suppress cardiac remodelling that lead to the onset of heart failure.
Targeted replacement of the cysteine finger of CR1, the entire CR2 domain and the ATP-binding site of CR3 generated A-Raf deficient mice on a C57BL/6 background. The mice developed at the expected Mendelian frequency and appeared normal at birth. By post-partum day (P) 2-3, however, they were noticeably smaller than littermate controls and showed a wasted appearance. By P5, their weight decreased to approximately 50% of that of their littermate controls and almost all A-Raf deficient mice died between P7 and P21. The few that survived to P21 appeared very wasted, had ruffled fur, a hunched appearance and were incapable of feeding once weaned. In addition, these animals suckled poorly and had less milk in their stomachs than normal littermate controls. Histological studies revealed an excessively reduced thymus, an anomaly of the colon and distinct neurological abnormalities such as abnormal movement and proprioception, abnormal position of their limbs at rest or when lifted by their tails, difficulties in maintaining an upright position, continuous tremor, rigidity of their musculature, and a distinct stress reaction with loss of bladder control and excessive agitation [Pritchard et al., 1996].
Interestingly, the phenotype of these A-Raf deficient mice was dependent on their genetic background. A-Raf deficient mice on a 129/SvEv background also displayed a lethal phenotype, while approximately 50% of the mice survived on a mixed C57BL76x129/Ola genetic background. All of the ’escapees’ were fertile and survived as runted animals for longer than 12 months. They did not develop intestinal problems and showed few of the neurological abnormalities described above. However, unlike normal littermates, they consistently drew their hind limbs into their bodies when lifted by their tails. The reason for the different phenotypes on a different genetic background remains unknown [Pritchard et al., 1996]. Finally, one group generated A-raf-∕-/c-Raf-1-∕- mice on a MF1 background. The vast majority of double KO mice died before they reached E10.5 and none survived to term. The double KO embryos were extremely small with truncated trails [Mercer et al., 2005b]. The complex phenotype of A-Raf KO mice corroborate the observations from in vitro studies that this kinase plays a pivotal role in various cellular processes.
Homozygous B-Raf KO breeding did not generate viable offsprings as all KO embryos died at E12.5. At E11.5, these embryos were significantly smaller than their WT or heterozygous littermates and exhibited extensive spontaneous apoptosis in liver, brain, heart and endothelial cells of the blood vessels. The reduced VEGF production probably resulted in the observed placental defects, which indicates that B-Raf plays a non-redundant role in embryonic and extraembryonic mammalian development [Galabova-Kovacs et al., 2006, Wojnowski et al., 1997].
Forebrain-specific B-Raf KO mice display deficits in hippocampal long-term potentiation and impairments in hippocampus-dependent learning and memory, including spatial learning and contextual discrimination. However, deletion of the B-raf gene did not disrupt other forms of learning and memory such as cued fear conditioning, conditioned taste aversion, basal synaptic transmission and paired-pulse facilitations. The mutants were neither impaired in motivation, motor coordination, nor vision. These results demonstrate that in the adult brain, hippocampal synaptic plasticity and hippocampus-dependent learning requires B-Raf [Chen et al., 2006].
Since the mutant B-RafV 600E shows a 500 times higher basal kinase activity than WT B-Raf protein and is commonly detected in human parathyroid carcinomas, transgenic mice expressing this mutant in a thyroid specific way, were generated. These mice showed enlarged thyroid glands by five weeks of age and a high prevalence of parathyroid carcinoma development compared to WT controls. Since these transgenic B-RafV 600E parathyroid carcinomas resemble their human counterpart in histopathological features, these mice can function as attractive models to test potential therapeutic strategies for the treatment of parathyroid carcinomas and confirm the key role of this oncoprotein in cancer [Knauf et al., 2005].
Transgenic mice on a C57BL/6 background with directed expression of B-RafV 600E died before birth: Abnormalities of spleen and liver, and bone marrow failure due to a reduced white blood cell production and evidence of non-lymphoid histiocytic neoplasia were observed. This indicates that the expression of RafV 600E in gestation is lethal to the embryo [Mercer et al., 2005a].
Homozygous c-Raf1-∕- embryos developed at the expected Mendalian frequencies, but 96% of the embryos on the inbred 129SvEms or mixed C57BL/6x129SvEms genetic background died between E10.5 and E12.5 and none survived past E13.5 and E16.5 respectively. In contrast, 67% of c-Raf1-∕- embryos on CD1 background were born alive, but died within hours after birth. Approximately 40% of c-Raf-∕- embryos on a MF-1 background survived after E16.5 and resulted in live births, surviving for up to one month. The embryos were much smaller, paler, and developmentally arrested. This arrest was particularly pronounced in the posterior region, which failed to complete axial rotation. Many embryos also exhibited a distended pericardium, irregular folding of the neural tube, and anomalies in the foetal liver. At E10.5, growth arrest of the placenta became detectable. In general, c-Raf1-∕- embryos had a thinner skin and less differentiated dermal and epidermal cell layers. Furthermore, the fusion of the eyelids, which occurs at E16 in WT mice, failed in 40% of the mutant embryos in the CD1 background and resulted in open eyelids at E18.5 or older [Mikula et al., 2001, Wojnowski et al., 1998].
Another study revealed that c-Raf-1 null embryos on 129OlaxC57BL/6 background appeared abnormal at E9.5. The embryos were smaller, developmentally arrested, anaemic and died before E12.5. All mutants lacked blood vessels in the yolk sac, had abnormal vascular formation, and increased number of apoptotic cells. On the 129 OlaxMF-1 background, however, embryos developed to term, but died within a few hours after birth. Again all embryos were lighter in body weight and anaemic. The livers of the KO mice contained fewer, but larger cells and there were also fewer areas of haematopoiesis compared to WT liver. The placenta were reduced and disorganized. Hüser and co-workers also generated TG mice expressing the non-phosphorylable mutant c-Raf-1DY 340F∕Y 341F [Hüser et al., 2001]. These mice, both on 129OlaxC57BL/6 and a 129OlaxMF-1 background, appeared normal in weight, behaviour, survival rate and T cell development. Approximately 40% of c-Raf1-∕- embryos on a MF-1 background survived after E16.5 and resulted in smaller mice (compared to the WTs), that lived for up to one month [Kamata et al., 2004, Mercer et al., 2005b]. In conclusion, the exact role of the Raf-1 protein remains elusive, but c-Raf-1 is essential for mouse development and plays a key role in preventing apoptosis. However, since its kinase activity seems indispensable for normal mouse development, c-Raf-1 may act more like a scaffold protein. Additionally, the genetic background of the mice affects the function of c-Raf-1, although the reason for this remains to be established.
Two studies have addressed the possible role of c-Raf-1 in cardiac development and physiology. Cardiac muscle-specific c-Raf -∕- mice in C57BL/6 background were born normally with no obvious phenotypical differences from WT littermates. The mice were fertile, developed to adulthood and exhibited a normal lifespan for up to 12 months of age. However, compared to c-Raf+∕+ mice, these conditional null mutants displayed cardiac dysfunctions and dilatation. c-Raf-1 has been shown to interact with ASK-1 and c-Raf-1 promotes cell survival by antagonizing ASK1. The double ASK-1-∕-/c-Raf-∕- KO rescued the histological and functional abnormalities observed in the cardiac muscle-specific c-Raf-∕- mice [Yamaguchi et al., 2004]. TG mice with cardiac-specific expression of dnRaf-1K375H appeared normal at birth, were fertile, and had normal cardiac structure and function. However, pressure overload provoked by TAC led to high lethality while all NTG mice survived [Harris et al., 2004]. These results indicate that c-Raf-1 is not essential for mouse heart development, but that normal cardiac function requires c-Raf-1.
Finally, epidermis-specific ablation of the c-Raf-1 gene did not affect the viability, fertility, and health of the mice. At four weeks of age, they displayed curled whiskers and a wavy fur, a phenotype lost after the first hair cycle. The architecture of the epidermis appeared indistinguishable from control littermates. However, epidermis-restricted Raf-1 deficient mice displayed delayed wound healing, probably due to a defect in keratinocyte migration [Ehrenreiter et al., 2005].
Lymphocyte-oriented kinase (LOK), predominantly expressed in the lymphoid organs, is a member of the STE/p21-activated kinase (PAK) family. LOKDexon1 deficient mice exhibited no obvious gross histopathological abnormalities and displayed no remarkable alterations in immune response [Endo et al., 2000].
Mekk1-∕- mice in a C56BL/6 background under non-stressed conditions are viable, fertile and phenotypically normal, except for the failure of eyelid closure, which results in post-natal inflammation of the eyes and blindness [Yujiri et al., 1998, Yujiri et al., 2000]. Compared to Mekk1+∕+ mice, Mekk1-∕- mice show an increased sensitivity to develop impaired cardiac function, myocyte apoptosis, and pulmonary congestion in response to pressure overload. Mekk1-∕- mice subjected to pressure overload displayed a higher mortality and lung/body weight ratio than the sham-treated controls [Sadoshima et al., 2002]. Blood vessels respond to damaging stimuli by activating a remodelling mechanism that leads to intimal hyperplasia. However, complete ligation of the right common carotid artery in Mekk1-∕- mice resulted in a significant decrease in intimal regions compared to WT mice. These results indicate that Mekk1 plays a role in vascular remodelling after blood-flow cessation [Li et al., 2005, Minamino et al., 2002].
Homozygous mice expressing kinase death Mekk1 (mekk1DKD∕DKD) are alive and fertile on a C57BL/6x129 background. However, on a C57BL6 background, the majority of the Mekk1DKD∕DKD mice survived beyond E14.5, but less than 2% of the mice survived to adulthood. All E14.5 mutant embryos studied, although morphologically normal, depicted an anaemic phenotype and showed defective erythropoiesis with accumulation of nucleated late erythroblasts, reduced number of foetal liver macrophages and very few mature red blood cells. The TG foetal liver showed reduced size and a hypocellular character compared to the WT foetal liver. Moreover liver necrosis as well as erythroplaki of the head and neck region were occasionally observed in these Mekk1DKD∕DKD embryos. No abnormalities in the morphology of placenta or differences in endothelial development was observed between Mekk1DKD∕DKD and WT mice. These studies indicate a role for Mekk1 in definitive erythropoiesis in the foetal liver [Bonnesen et al., 2005]. In accordance with Mekk1-∕- mice, all Mekk1-deficient (Mekk1DKD∕DKD) mice tested, were born with open eyes and the eyelid epithelium of the homozygous mutants was considerably thinner compared to WT embryos [Zhang et al., 2003a].
Homozygous MEKK2 deficient mice generated on a C57BL/6, as well as on a TC-1 genetic background lived approximately as long as WT mice and were fertile. In a non-stressed physiological setting, they appear healthy without overt developmental abnormalities. This contrasts with mekk2-∕- mice on a R-1 background, which were not viable. The basis for these genetic differences remains unclear [Garrington et al., 2000, Guo et al., 2002, Kesavan et al., 2004].
Mekk3+∕- mice displayed a normal phenotype and were fertile, although mekk3-∕- embryos died before E11.5. These embryos showed a disruption of blood vessel development throughout the embryo with a complete block in angiogenesis around E9.5. Furthermore, embryonic blood vessels in the placenta as well as the structure and integrity of the yolk sac vasculature developed abnormally. By E9.5, the mutant embryos were smaller than their WT littermates and by E10, the development of the heart was severely impaired [Yang et al., 2000, Yang et al., 2001]. Heart-specific expression of caMEKK3 in TG mice resulted in cardiac hypertrophy and sudden death [Abbasi et al., 2005]. These data demonstrate that blood vessel development during early embryogenesis and normal cardiac physiology requires functional MEKK3.
MEKK4-∕- mice on a mixed C57BL/6x129SvE background developed with decreasing Mendelian ratios from WT over heterozygotes to homozygotes. The few MEKK4-∕- mice that survived, exhibited a slightly reduced size at young age, but grew to adulthood without obvious defects [Chi et al., 2004]. However, MEKK4-∕- mice on a C57BL/6 background developed close to Mendelian ratios, but none survived at weaning age. Furthermore, more than 80% developed neuronal tube defects, including cranial exencephaly, spina bifida, curled tail, or any combination of these malformations. These defects were associated with increased apoptosis in the developing neuroeptihelium. These observations suggest that MEKK4, through MKK4, may protect against excessive cell death in the neuroepithelium thereby facilitating closure of the neural tube along the entire neuroaxis [Chi et al., 2005].
Similar phenotypical changes were observed in homozygous mice on a mixed C57BL/6X129Sv background expressing a kinase inactive MEKK4 variant (MEKK4K1361R). About 50% of the homozygous MEKK4K1361R embryos showed neural tube defects, associated with enhanced apoptosis in the neuroepithelium. Moreover, approximately 75% of the embryos had skeletal malformation and male homozygous MEKK4K1361R mice that survived to adulthood were infertile due to reduced sperm count and motility [Abell et al., 2005].
Mlk3 deficient mice in a C57BL/6J background were viable, healthy and lived out a normal life span, but displayed minor deficiency anomalies (reduced thickness of epidermal tissue) at the dorsal midline. The cause of this defect is unknown, but similar defects were registered in JNK-defcient mice. Currently, the in vivo role of MLK3 remains elusive [Brancho et al., 2005].
Some of the TG mice in a C57BL/6 background overexpressing WT MST1 from the heart-specific a-myosin heavy chain promoter exhibited heart failure and died prematurely as early as on day 15. These mice showed significant increases in LV end-diastolic and systolic dimension, significant decreases in LV ejection fraction, LV fractional shortening and LV wall thickness. The mice displayed dilation of all four cardiac chambers and mural thrombus formation in both atriums. Additionally, their lung weight/body weight and liver weight/body weight increased and their livers and lungs showed visible congestion at 3-4 months of age. The amount of apoptotic myocytes increased, which suggests enhanced cardiac myocyte death in the hearts of these animals. Moreover, these mice lacked compensatory cardiac myocyte hypertrophy. Transgenic dnMST1-miceK59R didn’t die prematurely nor did they show any signs of heart failure. However, these mice possessed diminished cardiac myocyte apoptosis and reduced size of myocardial infarction in response to ischemia/reperfusion. Both transgenic mice models suggest that MST1 is an important mediator of cardiac myocyte apoptosis in vivo [Yamamoto et al., 2003].
Mice lacking functional NIK possessed no peripheral lymph nodes, had defective B and T cells, and displayed impaired receptor activation of NFkB ligand-stimulated osteoclastogenesis. Compared to WT littermates, nlk-∕- mice showed significantly less periarticular osteoclastogenesis, less bone erosion, and they were completely resistant to antigen-induced, as well as genetic, spontaneous arthritis. This indicates an important role for NIK in the immune and bone-destructive components of inflammatory arthritis [Aya et al., 2005, Novack et al., 2003, Kortenjann et al., 2001b].
Transgenic nikD1-120nt mice on a 129/SvEv or mixed 129/SvEvXC57BL/6 backgrounds appeared normal in growth, behaviour, reproductive behaviour and nursing. However, they showed abnormal lymphorganogenesis (lack of peripheral lymph nodes and presence of defective B and T cells). Moreover, the spleen and thymus possessed abnormal morphology and poor immune responses upon immunization as indicated by increased susceptibility to bacterial eye infections [Yin et al., 2001].
Interestingly, in the alymphoplasia (aly/aly) mouse model, a naturally occurring NIK mutation (G855R) in a domain involved in the interaction with IKKa and TRAFs was identified. The phenotype of those mice resembles that of the nik KO mice (systematic absence of lymph nodes and Peyer’s patches, disorganized splenic and thymic structures, higher susceptibility to infections). Transgenic complementation with WT NIK restored the normal phenotype, confirming the implication of NIK in lymphorganogenesis [Shinkura et al., 1999].
Homozygous NLK-∕- mice on a C57BL/6 background, died in the third semester of pregnancy due to unknown causes. On a 129Sv background, however, mice survived for up to 6 weeks after birth. These mice displayed growth retardation, pronounced cerebellar ataxia, and severe compromised haemotopoiesis and aberrant differentiation of bone marrow stromal cells. In addition, an increased number of adipocytes, a reduced number of lymphoid cells and large blood sinuses were registered in these mice. The mean cellularity in spleen and thymus was also reduced by 90% and 70%, respectively [Kortenjann et al., 2001a].
TG mice with a B6DF/J background expressing dnPAK1K299R from the ovine BLG promoter, which is active during pregnancy and lactation, were normal during the virgin and early pregnant stages. At the late stages of pregnancy, the mammary glands of TG mice showed marked dystrophy with poorly developed alveoli and fewer visible branches compared to WT mice. The epithelial cells in the TG mammary gland exhibited a reduced proliferation rate and elevated apoptosis. Expression of PAK1K299R also reduced expression of the two major milk proteins b-casein and whey acidic protein [Wang et al., 2003]. The same group found that TG mice overexpressing the catalytic active mutant of PAK1T423E, but not age-matched WT mice, developed malignant mammary gland tumours and other breast lesions, including focal solid nodules, ductal hyperplasia, and mini-intraductal neoplasm and adenoma. Taken together, these findings suggest that PAK1 is required for alveolar morphogenesis and lactation function and involved in breast tumour progression [Wang and Tournier, 2006, Wang et al., 2006a].
Mutations of the human gene encoding PAK3 are associated with X chromosome-linked nonsyndromic mental retardation. PAK3 deficient mice on a C57/BL6x129Sc mixed background showed no apparent changes in viability, lifespan, fertility or locomotor activities, nor were deficits observed in neuronal structures and the actin cytoskeleton. However, compared to WT littermates, the PAK3-∕- mice were selectively impaired in late-phase hippocampal long-term potentiation and had accelerated extinction of the hippocampus-independent taste aversion associative learning task. These findings provide evidence that PAK3 deficiency leads to impaired synaptic plasticity and cognition. Therefore, these mice can be used as a model for studying nonsyndromic mental retardation [Meng et al., 2005].
While PAK4+∕- mice appeared normal and were fertile, none of the PAK4-∕- embryos on a C57BL/6 background survived beyond E10.5. Lethality was due to a heart defect and abnormal neuronal development: thinner myocardial walls and a dilation of the atrium and sinus venosus characterized the heart defect, while neuronal abnormalities included thin neuroepithelium around the hind- and forebrain, lack of neurite outgrowth, defect in neuronal migration, defect in motor neuron differentiation and migration, impaired development of ventral interneurons, and improper folding or pinching of the caudal end of the neural tube. The molecular basis for cardiac and neural deffects is not yet known, but the absolute requirement of PAK4 for normal development suggests that the other PAK members cannot compensate for the functions of PAK4 [Qu et al., 2003].
PAK5 is highly expressed in the eye and the brain, specifically in neurons, but is also expressed at lower levels in several other tissues. In an effort to unravel the biological function of this kinase, Li and Minden generated homozygous PAK5 deficient mice on a C57BL/6 background. These KO mice were born at Mendelian frequencies, were fertile, and developed normally without any obvious phenotype. The expression levels of PAK1, PAK2, PAK4 and PAK6 were unaffected compared to WT mice. These findings suggest a functional redundancy between PAK5 and other PAK members, especially PAK6 which is also brain enriched. The development of PAK6 and PAK5/PAK6 double KO mice may help to establish possible redundancy between both kinases and determine their biological role [Li and Minden, 2003]. Since phenotypes can vary with different genetic background, studies with PAK5 KO mice in different backgrounds may also shed light on the function of this protein.
Heterozygous TG mice on a FVB/NxICR genetic background that express an activated TAK1 appeared normal at birth, but all died within 2 weeks. At 9-11 days, cardiac lung and liver mass of all TG mice had increased compared to control littermates. At this age, the hearts revealed hypertrophic myocytes with hyperchromatic nuclei and increased apoptosis, myocyte disorganization, interstitial fibrosis, and impaired sys- and diastolic function. These results indicate that dysfunction of TAK1 may elicit myocardial hypertrophy and heart failure. However, whether endogenous TAK1 is required for normal cardiac functions cannot be firmly concluded from this model, therefore a cardiac-specific Tak1 gene disruption may be more informative [Zhang et al., 2000]. Studies with TAK1-∕- embryos on a C57BL/6 or a mixed 129xC57BL/6 background revealed severe abnormalities of the neural tube and embryonic mortality around day E10.5 with none surviving beyond E12.5 [Sato et al., 2005, Shim et al., 2005].
TG mice with tissue/organ-specific ablation of normal TAK1 function were generated to identify the biological role of this kinase in certain physiological processes. Studies with mice containing a B cell-specific TAK1 deficiency suggested that TAK1 is responsible for the development of B-1 B cells and for the induction of humoral immune responses [Sato et al., 2005]. Mice with keratinocyte-specific TAK1 deficiency, as well as mice with keratinocyte-specific expression of a kinase death TAK1 variant were born at the expected Mendelian ratios and were grossly indistinguishable from WT littermates from birth until postnatal day 2-3. By postnatal day 7, however, the mutant mice displayed hard inflexible skin and widespread scaling, and diseased lips that may have affected nursing. All mutant mice died between postnatal days 7 and 8. Histological analysis of the skin of these mice revealed a progressive epidermal condition involving severe apoptosis, hyperkeratosis, inflammation and eventually epidermal erosion. These data demonstrate that TAK1 is essential for skin homeostasis [Sayama et al., 2006, Omori et al., 2006].
Mice lacking TAK1 in T cells are viable, but exhibited a significant reduction of CD4+ and CD8+ single-positive thymocytes in the peripheral tissues. The defective development of these T-cells was due, at least in part, to increased apoptosis of these cells. The exact role of TAK1 in thymocyte development remains to be determined, but the results extend the pivotal role of TAK1 in both innate and adaptive immunity [Liu et al., 2006].
While MEK1+∕- heterozygous mice developed normally, homozygous MEK1 KO mice did not yield viable offsprings. Histological analysis of E10.5 WT and MEK1-∕- yolk sacs revealed that MEK1 plays an essential role in placental development. MEK1 mutant yolk sacs displayed a reduced number of blood cells, distended blood vessels, a less well-defined spongiotrophoblast layer and a more compact labyrinthine region. One third of the homozygous mutant embryos were smaller than their WT and heterozygous littermates, and some showed haemorrhaging, which suggests anomalies in blood circulation or blood vessel formation. Moreover, E10.5 MEK1-∕- embryos exhibited necrosis in various tissues [Giroux et al., 1999].
TG mice with cardiac-restricted expressions of an activated MEK1 in the heart demonstrated concentric cardiac hypertrophy with an approximate 50% increase in septal thickness and LV posterior wall thickness, without signs of cardiomyopathy or lethality (for up to 12 months of age). Additionally, MEK1 TG hearts showed resistance to ischemia/reperfusion-induced apoptosis [Bueno et al., 2000]. Expression of activated MEK1 in the lens disrupted the expression of a glucose transporter and of the crystalline protein associated with differentiation. The increased glucose levels in TG lenses induced cataract formation [Gong et al., 2001]. A TG mouse model of skin-restricted MEK1 expression showed a pronounced epidermal hyperproliferation and hyperkeratosis. Some older mice also developed papillomas at sites of wounding, including the tail, lower back and ears [Hobbs et al., 2004].
Mek2-∕- progeny developed according to Mendelian ratios. These KO were viable, fertile and showed no evident growth defects and morphological alterations, rendering MEK2 indispensable for normal mouse growth and development. Surprisingly, while MEK1+∕-, as well as MEK2+∕- heterozygous mice developed normally, 85% of the MEK1+∕-/MEK2+∕- double heterozygous mice died before birth. This indicates that the absence of one allele of each mek gene had more deleterious effects on mouse development than the absence of both mek2 alleles. This phenotype also suggests that MEK2 contributes to embryogenesis, but its exact role remains elusive [Belanger et al., 2003].
Mek3 KO mice were viable and without any obvious morphological or histological defects. The KO mice contained normal numbers of thymocytes and splenocytes, the major cell surface markers of T and B lymphocytes and normal bone marrow derived dendritic cells Lu et al. [1999]. Additionally, some researchers noted defects in T-cell function and cytokine production [Beardmore et al., 2005, Kaiser et al., 2004]. Cardiac-specific transgenic mice expressing dnMEK3 displayed enhanced cardiac hypertrophy following aortic banding, angiotensin II infusion and phenilephrine infusion for 14 days [Braz et al., 2003].
MEK4-∕- embryos were severly aneamic and died between E10.5 and E12.5. Histological analysis of all embryonic organs revealed normal appearance and overall morphology, except for the liver, that was severely disorganized and contained significantly reduced numbers of parenchymal hepatocytes. Importantly, liver remnants from MEK4-∕- embryos contained haematopoietic precursor cells and large clusters of erythroid cells, indicative of hepatic erythropoiesis [Ganiatsas et al., 1998, Yang et al., 1997a, Nishina et al., 1999].
Heterozygous MEK5 KO mice appeared healthy and fertile. In contrast, homozygous embryos died at approximately E10.5. At this stage, MEK5-∕- embryos were smaller than WT or heterozygous littermates and displayed retarded development of the head, limbs and heart [Wang et al., 2005].
Homozygous MEK6 KO mice were viable, fertile and displayed no developmental or histological abnormalities. The KO mice contained a comparable number of thymocytes and splenocytes with similar expression of the major surface markers, as WT mice [Beardmore et al., 2005, Kaiser et al., 2004, Tanaka et al., 2002]. Skeletal preparations of TG mice that express a constitutively active MEK6 revealed a shortened axial and appendicular skeleton, which resulted in a dwarf phenotype [Zhang et al., 2006]. TG mice with cardiac-specific dnMEK6 showed features similar to dnMKK3 mutants and enhanced cardiac hypertrophy [Braz et al., 2003]. Double KO of MEK3-∕- and MEK6-∕- mice resulted in embryonic lethality due to placental and vascular defects [Brancho et al., 2003, Adams et al., 2000, Beardmore et al., 2005].
MEK7-∕- mice died during embryogenesis due to unidentified causes so far [Dong et al., 2000].
The ERK1-deficient mice were viable, fertile, of normal size and displayed no histological abnormalities. The numbers and percentages of splenic CD4+ and CD8+ T-cells, B cells, and macrophages resembled those of WT mice. Additionally, these mice exhibited unaltered antigen-specific proliferation, cytokine production and clonal sizes of CD4+ T cells. This indicates that ERK1 is not critical for the maturation, activation and differentiation of T cells and their effector function in the periphery. Nevertheless, Pages et al. showed a reduction in the number of mature CD4+CD8- and CD4-CD8+ single-positive thymocytes, which could be explained by the fact that both studies targeted different parts of the gene [Ferguson et al., 2006, Pages et al., 1999, Nekrasova et al., 2005]. Histochemical analysis of the brain revealed no anatomical differences between KO mice and littermate controls. However, behavioural tests showed a statistically significant increase in locomotor activity of ERK1-∕- mice. Furthermore, ERK1 also plays a role in the formation of striatum-dependent long-term memory and auditory sensory memory but is not necessary for hippocampus- and amygdala dependent emotional learning, as electrophysiological and behavioural evidence indicated. ERK1-deficient mice displayed an increased conditioned place preference to morphine and cocaine [Umbricht et al., 2004, Mazzucchelli et al., 2002, Ferguson et al., 2006, Selcher et al., 2001]. Furthermore, these mice also showed decreased adiposity: they are resistant to obesity when challenged with a high-fat diet and are protected from insulin resistance [Bost et al., 2005].
ERK1 deletion resulted in the exacerbation of progression and severity of experimental autoimmune encephalomyelitis (EAE), ncreased number of infiltrating cells and myelin destruction was observed in the spinal cord of ERK1-∕-mice [Agrawal et al., 2006].
Two different phenotypes of ERK2-deficient mice were obtained in two different backgrounds. In C57BL/6 mice, deletion of the ERK2 locus led to embryonic lethality before E8.5. However embryos developed according to Mendelian frequencies at E6.5 and E7.5. The ERK2-∕- embryos were significantly smaller than their WT or heterozygous littermates. Mutant embryos at E6.5 showed an abnormal morphology beginning oval in shape, with no obvious proximodistal and anterioposterior polarities, which could be easily distinguished in WT embryos at this stage. ERK2-∕- embryos were characterized by failures in the development of the extra-embryonic ectoderm and ectoplacental cone [Yao et al., 2003, Saba-El-Leil1 et al., 2003]. These morphological defects in ERK2-∕- embryos could result from primary defects at the level of the polar trophoectoderm. Homozygous ERK2 KO mice also displayed placental abnormalities [Hatano et al., 2003]. ERK2-∕- embryos on a BALB/C background, died at E6.5 and had difficulties in mesodermal differentiation [Yao et al., 2003]. These studies indicate that absence of ERK2 leads to impaired placental and trophoblast formation and mesoderm differentiation [Aouadi et al., 2006].
Homozygous ERK5 KO embryos survived up to E9.5 and E10.5 but all underwent resorption or died beyond E11.5. The cause laid in impaired placental and blood vessel development. The basic placental structures could form in the absence of ERK5, but the thickness of the labyrinthine layer was reduced due to apoptosis. Additionally, the KO embryos showed retarded growth and abnormal morphology, especially in the head and lower trunk regions. ERK5 also plays an essential role in cardiac development as the myocardium wall of E9.75 ERK5-∕- embryos was thinner than in WT embryos. Addtionally some bleeding was seen in a proportion of ERK5-∕- embryos [Yan et al., 2003, Aouadi et al., 2006].
The first JNK1 KO mice generated, seemed fertile, of normal size and appeared to have a normal lymphocyte development, without further studies done on thymocyte selection [Dong et al., 1998]. In transgenic mice expressing dnJNK1T183A,Y 185F and additional cross breeds of dnJNK1/AP1 luciferase or dnJNK1/Cyt c TG mice, the absence of JNK1 didn’t seem to affect positive selection, but resulted in defective deletion of double positive CD4+CD8+ thymocytes [Rincon et al., 1998b].
Similarly, single JNK2 KO mice developed normally, were fertile, showed no overt developmental abnormalities, and T and B lymphocyte differentiation and apoptosis appeared normal as well. However, JNK2 KO mice displayed impaired activation of peripheral CD4+ T-cells, attributed to a defect in IFNg early in differentiation. [Sabapathy et al., 1999a, Yang et al., 1998, Aouadi et al., 2006].
In order to investigate possible redundancy between JNK1 and JNK2, a double homozygous KO was constructed. JNK1 KO and JNK2 KO mice were used to investigate their role in rheumatoid arthritis. The study revealed that absence of JNK1 and 2 suppresses MMP and bone destruction. However, it reported no particular behavioural phenotyping [Han et al., 2001]. In another study, Dong et al. crossbred a T-cell specific KO of JNK1 with a JNK2 KO and generated a Rag-JNK1-∕-/JNK2-∕- TG mouse line. In both cases, mice develop normally, were fertile, showed normal T and B lymphocyte development but increased production of Th2 cytokines (IL-2). This suggests that JNK1 and 2 regulate Th-cell differentiation by potentiating Th1 responses via inhibition of Th2 cytokine production [Dong et al., 2000, Yang et al., 1998, Aouadi et al., 2006]. Finally, since Drosophila homologues for the JNK signalling pathway are required for dorsal closure of Drosophila embryos, JNKs could play a role in mammalian development as well. To investigate this, Kuan et al. generated KO mutants of JNK1/2, JNK1/3, JNK2/3. JNK1/3 KO and JNK2/3 KO progenies developed according to Mendelian ratios, whereas the JNK1/2 KO mutants died between E11 and E12. These embryos showed a considerable hind brain neural tube defect, which indicates that JNK1 and 2 play an essential role in the morphogenesis of the mammalian brain, a role for which JNK3 cannot substitute [Li et al., 2004, Kuan et al., 1999, Sabapathy et al., 1999b, Aouadi et al., 2006].
Pressure overload of the heart caused no differences in trans stenotic pressure gradients in JNK1-∕-, JNK2-∕-, JNK3-∕- nor WT mice. Cardiac hypertrophy induced by 7D TAC and cardiac function in these TG mice resembled the responses of the WT mice. This indicates a less important role for JNKs in inducing cardiac growth after pressure overload. However, for JNK1-∕- mice cardiac function deteriorated after TAC: they showed preserved LV end diastolic dimensions but displayed increased LV end systeolic dimensions. However, after the acute deterioration, the cardiac function improved mildly until it was indistinguishable from WT mice after 12 weeks. This indicated that JNK1 is selectively required to promote survival signals and to preserve cardiac function in acute response to cardiac pressure overload [Tachibana et al., 2006].
Tuncman et al. intercrossed JNK1-∕- and JNK2-∕- mice and subjected them to both regular and high fat diets. JNK1-∕- mice showed reduced obesity and systemic insulin resistance, an effect absent in JNK2-∕- mice. This suggests that obesity-induced insulin resistance is mainly mediated by JNK1. This study indicates that both JNKs play important roles in the regulation of lipid and glucose metabolism in the whole organism, although the role of JNK2 is less obvious, due to crosstalk between both JNKs [Tuncman et al., 2006, Aouadi et al., 2006].
Next, the role of JNKs in liver pathology was examined. Toxin-induced liver injury was markedly reduced in JNK2 KO mice compared to JNK1 KO and WT mice, suggesting that the absence of JNK2 protects from liver injury and associated mortality [Gunawan et al., 2006, Wang et al., 2006b]. A study by Sakurai and coworkers showed that JNK1 KO mice were much less susceptible to chemical-induced hepatocellular carcinoma than WT animals. The tumours derived from JNK1-∕- mice exhibited lower proliferation rates than tumours from WT mice. Moreover, large tumours with neovascularization were observed in WT but not in JNK1 KO mice. In conclusion, these data point to a role for JNK1 in tumour progression (proliferation and neovascularization) as well as initiation or early tumour promotion [Sakurai et al., 2006]. Finally, JNK1-∕-and JNK2-∕- mice placed on a methionine- and choline-deficient diet developed fewer cases of steatohepatitis, compared to WT mice. The same diet also lead to reduced lipid accumulation in the liver of JNK1 KO mice, compared to JNK2 KO mice and WT littermates. Therefore, the authors believe that JNK not only modulate acute states of injury and cell death but also promote chronic tissue injury. [Schattenberg et al., 2006].
Homozygous JNK3D211-267 mice displayed normal size, fertility, normal histological appearance, and normal development. However, JNK3 KO mice showed remarkable resistance when injected with an epileptogenic dose of kainic acid, which contrasted to the reaction of JNK1 KO and JNK2 KO. This indicates JNK3 could be used as a therapeutic target for a wide range in neurological disorders (including ischemia and neurodegenerative diseases) that result from neurotoxicity [Yang et al., 1997b, Aouadi et al., 2006].
p38a null mice in several different backgrounds died in utero because of a lack of oxygen and nutrients due to defects in placental angiogenesis. After E11.5, KO embryos were detected less frequently and decreasing when mice were backcrossed from (129/Svx129/J)F1xC57BL/6 into a C57BL/6 background. Of the offsprings that survived, the heterozygous progeny was fertile, but the homozygote mice were not. The TG mice had a slight pallor and contained smaller and paler foetal livers, and a normal yolk sac with few circulating erythroycytes. By using markers for hematopoietic cell types and developmental stages, the study revealed a deficiency in the expansion and differentiation of CFU-E progenitors, which could be corrected. This indicates that erythroid differentiation requires p38a. Additionally Epo expression seems to require p38a as well [Tamura et al., 2000, Adams et al., 2000, Mudgett et al., 2000].
To circumvent the lethality of homozygous p38a-∕-mice, thereby allowing the examination of the putative role of p38a in different physiological processes, heterozygous TG mice expressing dnp38a or mouse models with tissue-specific depletion of p38a have been generated.
Several groups investigated the role of p38a in cardiac function. Kaiser et al. generated a mouse model with cardiac specific deletion of both p38a alleles. They report that 6-8 weeks old mice showed reduced cardiac function which seemed more severe in a C57BL/6 background, compared to FVB/N [Kaiser et al., 2004]. In another study, 8-10 weeks old heterozygous p38a and p38b mice were used. In contrast to p38b mice, but similar to the former study, p38a mice showed an increased survival and resistance to myocardial infarction with no further abnormalities in body and heart weight, heart architecture, dia- and systolic blood pressure and LV pressures [Otsu et al., 2003, Braz et al., 2003] . The role of p38a in hypertrophy and fibrosis as a response to cardiac ischemia was addressed by construction of cardiac-specific p38aD40-83 floxed mice in C57BL/6 background. These KO mice appeared normal and were externally indistinguishable from WTs. They showed normal cardiac structure and function (echocardiography, cardiac caterization), but revealed different responses to biomechanical stress (pressure overload) compared to WT littermates. The authors suggested that the kinase is essential for survival but not hypertrophic growth in the heart [Nishida et al., 2004].
Other studies with transgenic mice expressing dnp38T180A∕Y 182F mice revealed increased LV ejection and reduced end dia- and systolic volumes compared to the NTG [Ren et al., 2005]. After infarction, LV mass and mass index decreased in dnp38a compared to NTG mice. Therefore, the authors concluded that inhibiting p38a during the prolonged phase of remodelling may be beneficial for patients. As such, ventricular remodelling is suited for medical intervention because it occurs in days and weeks following myocard infarct. On the other hand, a study with p38aDT180A∕Y 182F mice in FVB/N background reported that cardiac hypertrophy already developed at baseline, and increased after abdominal aortic occlusion, as was measured by heart to body weight ratios. The effect was also visible in MEK3-∕-mice, but absent in MEK6-∕-mice. In the end, both the MEK3-∕-mice and the p38aDT180A∕Y 182F mice succumbed to cardiomyopathy [Braz et al., 2003]. In another study, p38aT180A∕Y 182F mice and additionally p38b mice (both in Black Swiss background), displayed hypertrophic hearts in both mouse models after constriction of the aorta and carotid arteries. Furthermore, the hearts of these mice showed no evidence of fibrosis, leading the authors to conclude that cardiac fibrosis is not always associated with cardiac hypertrophy [Zhang et al., 2003c].
To determine if p38a is predominantly involved in the progression of inflammatory disease in a Contact Hypersensitivity model (type IV hypersensitivity), and whether topical application of p38 inhibition can be used as therapeutic utility, Takanami-Ohnishi et al. compared the responses of 2.5-Dinitro-1-Fluorobenzene (DFNB) induced hypersensitivity in WT and p38a+∕- in the presence or absence of p38 inhibitor. Ear swelling and infiltration of inflammatory cells (in ear skin and lung) were reduced in p38a+∕- compared to WT mice. Additionally, the expression of cytokines was altered. The authors concluded that targeting p38 in hypersentivity models could be used by therapeutic strategies [Takanami-Ohnishi et al., 2002].
Another study investigated dnp38aT180A∕Y 182F expressed under the lck promoter in TG mice in a B10Br background to determine possible effects on the immune system. These mice resembled WT mice in thymus development, and proliferation and homeostasis of lymphocytes in the periphery. However, the activation of Th1 cells was impaired in dnp38 TG mice. This indicates that inhibition of p38a in CD4+ T-cells causes impairment of Th1 responses but doesn’t seem required for Th2 cell response [Rincon et al., 1998a].
Finally, to investigate the relation between p38a deficiency and the risk of renal insufficiency, p38a heterozygous mice were constructed. These transgenic mice weighed approximately the same as WT litters, they appeared normal, and had normal urine biochemical parameters. However, the kidney to total body weight had increased at 21 weeks of age and the p38a+∕- mice showed increased water intake which became significant at 21 weeks of age (polydipsia). Histological abnormalities became more pronounced over a time course from 4 to 21 weeks, resulting mainly in dilated proximal tubules and showing vacuolar degeneration. Furthermore, these renal abnormalities were more severe in males compared to females. The authors concluded that one p38a allele is insufficient for normal renal structure and functional integrity [Maruyama et al., 2003].
p38b KO mice were born at expected frequency, had normal fertility, normal thymocyte development, normal size and no apparent health problems or phenotype. To investigate possible roles in inflammatory diseases, p38b-∕-mice were crossbred to TNFDARE mice, which are more prone to develop these type of diseases. The resulting progeny didn’t differ from the controls suggesting p38b plays no critical role in inflammatory diseases [Beardmore et al., 2005]. Another group demonstrated that p38b deficiency did not affect basal nor insulin-mediated glucose uptake, which indicates that p38b doesn’t participate in the hormonal activation of glucose transport [Turban et al., 2005].
The role of p38b in heart functions was also investigated. Here, heterozygous p38b+∕-mice didn’t show, in contrast to p38a+∕- mice, an increased survival and resistance to myocardial infarction [Braz et al., 2003, Otsu et al., 2003]. TG mice crossbred from dn14-3-3 and cardiac specific forms of dnp38a or dnp38b revealed that p38b plays a more important role than p38a in the cardiomyocyte survival in response to pressure overload: single dn14-3-3 mice were intolerant to pressure overload and died, whereas in dn-14-3-3/dnp38b and in p38a/dn14-3-3 mice respectively 100% and 60% survived [Zhang et al., 2003b].
Single as well as double KO models for p38g and p38d are viable, fertile and appeared normal, but no further information is available on their phenotype [Beardmore et al., 2005, Sabio et al., 2005, Aouadi et al., 2006].
Ueda et al. investigated the role of Mnk1 and Mnk2, by constructing KO mouse models of Mnk1 and Mnk2. Both single and double KO mice were viable, fertile and displayed no apparent abnormalities, in contrast to Drosophila, where Mnk1 and Mnk2 are required for cell growth and ontogenic development [Ueda et al., 2004, Reiling et al., 2005].
In order to better understand the cause of drug abuse due to the induction of immediate early genes in the striatum, MSK1-∕- mice were constructed. These mice exhibit no apparent health problems and displayed normal size, weight, fasting glucose levels, T-cell and brain development and overall anatomy (including specific brain regions involved in addiction). Additionally, the mice showed normal habituation to a new environment as well as normal horizontal and vertical activity throughout the light/dark cycle. However, locomotor sensitization, which occurs after repeated cocaine exposure, seemed to require MSK1 as: the KO mice displayed no enhanced locomotion, compared to littermate controls. Furthermore, as the conditioned controls also showed increased preference for cocaine compartments in high dosage circumstances, this response was absent in the KOs although they displayed a similar but milder behaviour during low dosage conditioning [Brami-Cherrier et al., 2005].
Single and double KO of MSK1 and MSK2 were viable and fertile, without any obvious health problems, but no behavioural information is available [Wiggin et al., 2002, Casanova et al., 2002].
In general, MK2 deficient mice were fertile, viable, of normal size and depicted no specific behavioral defects, nor did these mice show chromosomal alignment defects and spindle abnormalities during mitosis, as reported in MK2-deficient Drosophila flies. However, MK2-deficient mice demonstrated an increased sensitivity to high-salt feeding and resistance to LPS induced shock. Additionally, they produced reduced amounts of cytokine in spleen cells and serum [Gaestel, 2006, Kotlyarov et al., 1999]. This indicates that MK2 provides a unique target for anti-inflammatory therapy [Shi et al., 2003].
In ischemia models for both heart and brain, the MK2 KO mice showed reduced infarct size. In the heart model, MK2 KO mice differed in baseline cardiac function, coronary flow and heart rate. Furthermore, they showed a markedly decreased amount of apoptosis, and a better recovery. In the brain ischemia model, physiological and apoptotic parameters, gliosis or microglial activation appeared normal, but the mean arterial blood pressure increased. These KO-mice showed an improvement in motor function as well [Shiroto et al., 2005, Wang et al., 2002]. Culbert et al. isolated microglia from MK2 deficient mice in order to investigate its role in Alzheimer’s disease, by means of cellular studies. By consequence, there is no behavioural data available [Culbert et al., 2006].
MK2 deficient mice in a DBA/ILacJ background rendered the mice susceptible to collagen induced artritis (CIA). The joints of MK2-∕- and MK2+∕- mice displayed reduced severity and incidence of artritis, in contrast to WT littermates of whom none had normal joints. The MK2-∕- mice had only minimal pannus formation or fibrillation of articular cartilage, while the MK2+∕- mice displayed intermediate severity. Additionally, the MK2 deficient mice expressed reduced levels of IL-6 and TNFa, known to be involved in rheumatoid arthritis (RA). This makes MK2 and ideal target to ameliorate numerous inflammatory diseases [Hegen et al., 2006]. The same cytokine profile was found in the pancreas of the MK2-∕- mice. In this study pancreatic oedema and subsequent acute pancreatitis was induced by injecting the mice with cerulein [Tietz et al., 2006]. These data confirm the role of MK2 in inflammatory diseases.
Finally, MK2 and MK5 double KOs were viable, but
produced slightly fewer
TNFa compared
to WT littermates. This indicates that MK3, for which no
mouse model exists yet, may compensate partially for the absence of
MK2 [Shi
et al., 2003].
~> in the
meanwhile MK3 has gotten a KO mouse model which illustrates that MK3
can partially overcome a lack of MK2! see Molecular and Cellular
Biology 2007,27(1):170-181!
Depending
on the background of the mice, MK5 KO
mice develop deficits [Gaestel, 2006].
MK5 deficient mice in 129xC57BL/6 background are fertile, viable, and
showed no
behavioural abnormalities, or changes in morphology of the tissues
(especially the heart,
skeletal muscle and pancreas), or differences in LPS-induced endotoxic
shock,
compared to WT mice. The same MK5 KO mice in C57BL/6 background
revealed
embryonic lethality with incomplete penetrance, with only 50% of the
homozygous
MK5 mutants surviving after E12. This indicates that the background
plays a
considerable role in the phenotypical consequences of the deletion of
the kinase. The
viable homozygous MK5 mutants show no morphological/histological
abnormalities
over a time course of 3 to 24 weeks, although they start out smaller in
size.
The maternal placenta of hemizygous MK5 mice showed no abnormalities
either [Schumacher et al., 2004].
~> in the meanwhile, a new
report appeared which indicated that MK5 deficiency enhanced
DMBA-induced skin papillomas. The model knocked out a different
exon compared to the original KO mouse model
(Cell,2007;128:295-308.Sun et al :-))
Viral (e.g. influenza A virus, Coxsackie B3 virus, adenovirus, herpesviruses, HIV), as well as protozoal (e.g. Leishmania species) and bacterial (e.g. Mycobacterium leprae) infections can modulate the activity of MAP kinases in cell cultures [Ludwig et al., 2003, 2006, Schumann and Dobbelstein, 2006, Tapinos and Rambukkana, 2005, Sumbayev and Yasinska, 2006, Olivier et al., 2005, Brinkmann and Schulz, 2006]. However, the role of MAP kinases for successful in vivo replication of these parasites is less well established. Ölschläger and colleagues examined the in vivo relevance for increased influenza virus A replication in cells with an upregulated Raf/MEK/ERK signalling pathway. Therefore, they constructed TG mice with a lung specific expression of an activated c-Raf-1 mutant. Compared to WT mice, influenza A virus infection in TG mice resulted in more severe clinical symptoms and increased mortality. However, they observed no alteration in the immune responses of the TG mice, thus ruling out that the increased viral replication in TG mice was an indirect effect of an impaired immune response against the virus [Olschläger et al., 2004]. The requirement of Raf/MEK/ERK pathway for efficient in vivo influenza A virus replication illustrates that transgenic mice expressing activated or dominant negative forms of MAP kinases may be valuable models to determine the role of a particular MAP kinase in pathogen propagation in vivo and for testing the potentials of specific MAP kinase inhibitors as anti-pathogenic therapy. A prerequisite for the use of specific MAP kinase inhibitors is that no resistant strains emerge.
The mitogen-activated protein kinases are important signalling molecules that participate in different cellular events and are potential targets for intervention in inflammation, arthritis, heart failure, cancer, neurological degeneration, and other diseases. There are many synthetic blockers of MAP kinase which possess potent anticancer activity or anti-inflammatory [Milella et al., 2002, Sebolt-Leopold and Herrera, 2004, Kohno and Pouyssegur, 2006, Mikalsen et al., 2006, Revesz et al., 2004]. The MAP kinase TG mice can be useful to validate MAP-kinase inhibitors to treat diseases. Indeed, several p38 MAP kinase inhibitors were effective in a murine model of collagen-induced arthritis and they prevented progression of the disease [Badger et al., 1996]. They also had protective effects in antigen-induced arthritis [Badger et al., 2000], or showed anti-inflammatory effects [Wada et al., 2005].
TG mouse models have been proven to be valuable tools to identify the biological role of MAP kinases. The phenotypical changes observed in such mice have in a few cases (e.g. B-Raf, PAK3) confirmed the implication of MAP kinase in human cancers or in human genetic diseases. However, the interpretation of an observed phenotype in a TG mouse and the extrapolation to the biological role of the kinase in humans should proceed with caution. In this review, we discussed several examples in which different genetic backgrounds of TG mice result in different phenotypes. The reason for these differences remains unclear. In this regard, studies that used mice in mixed backgrounds may be more representative for the human population, compared to homozygous mice. In any case, experiments performed in mice with distinct genetic backgrounds, as well as overexpression of dominant negative or constitutive active variants may further increase our knowledge on the genuine role of MAP kinases.
Some MAP kinase TG mice did not show any obvious phenotype under normal living conditions. Many MAP kinases belong to families with high homology between the different members and overlapping expression patterns in the body which might explain the apparent dispensability of a particular MAP kinase (redundancy). The use of double or even a multiple KO approach may help to solve the possibility that different members can substitute for each other. Alternatively, the kinase may exert a special function under specific circumstances which have not been approached nor recognized in a particular study.
There exists no doubt that transgenic mice models have enormously expanded our knowledge of the MAP kinases and that these mice form excellent alternatives to test novel drugs against MAP kinases designed in the fight against pathogenic infections and human diseases. Despite elaborate, time consuming and expensive to generate, TG mice will still constitute important tools in basic and applied research in the future.
S. Abbasi, B. Su, R.E. Kellems, J.H. Yang, and Y. Xia. The essential role of MEKK3 in angiotensin II-induced calcineurin/nuclear factor of activated T-cells activation. Journal of Biological Chemistry, 280(44):36737–36746, 2005.
Amy N. Abell, Jaime A. Rivera-Perez, Bruce D. Cuevas, Mark T. Uhlik, Susan Sather, Nancy L. Johnson, Suzanne K Minton, Jean M. Lauder, Ann M. Winter-Vann, Kazuhiro Nakamura, Terry Magnuson, Richard R. Vaillancourt, Lynn E. Heasley, and Gary L. Johnson. Ablation of MEKK4 kinase activity causes neurulation and skeletal patterning defects in the mouse embryo. Molecular and Cellular Biology, 25(20):8948–8959, 2005.
Ralf H. Adams, Almudena Porras, Gema Alonso, Margaret Jones, Kristina Vintersten, Simona Panelli, Amparo Valladares, Lidia Perez, Ruedinger Klein, and Angel R. Nebredda. Essential role of p38a MAP kinase in placental but not embryonic cardiovascular development. Molecular Cell, 6(1):109–116, 2000.
A. Agrawal, S. Dillon, T. L. Denning, and B. Pulendran. ERK1-∕- mice exhibit Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis. The Journal of Immunology, 176(10): 5788–5796, 2006.
M. Aouadi, B. Binetruy, L. Caron, Y. LeMarchand-Brustel, and F. Bost. Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie, Epub ahead of print, 2006.
Jonathan D. Ashwell. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nature Reviews Immunology, 6(7): 532–540, 2006.
Kunihiko Aya, Muhammad Alhawagri, Amanda Hagen-Stapleton, Hideki Kitaura, Osami Kanagawa, and Deborah Veis Novack. NF-kB-inducing kinase controls lymphocyte and osteoclast activity in inflammatory arthritis. Journal of Clinical Investigation, 115(7):1848–1854, 2005.
A.M. Badger, J.N. Bradbeer, B. Votta, J.C. Lee, J.L. Adams, and D.E. Griswold. Pharmacological profile of SB203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock, and immune function. Journal of Pharmacology and Experimental Therapeutics, 279(3):1453–1461, 1996.
A.M. Badger, D.E. Griswold, R. Kapadia, S. Blake, B.A. Swift, S.J. Hoffman, G.B. Stroup, E. Webb, D.J. Rieman, M. Gowen, J.C. Boehm, J.L. Adams, and J.C. Lee. Disease-modifying activity of SB242235, a selective inhibitor of p38 mitogen-activated protein kinase, in rat adjuvant-induced arthritis. Arthritis and Rheumatism, 43(1):175–183, 2000.
Victoria A. Beardmore, Heather J. Hinton, Christina Eftychi, Maria Apostolaki, Maria Armaka, Joanne Darragh, Joanne McIlrath, Julia M. Carr, Laura J. Armit, Carol Clacher, Loraine Malone, George Kollias, and J. Simon C. Arthur. Generation and characterization of p38b (MAPK11) gene-targeted mice. Molecular and Cellular Biology, 25(23):10454–10464, 2005.
L.F. Belanger, S. Roy, M. Tremblay, B. Brott, A.M. Steff, W. Mourad, P. Hugo, R. Erikson, and J. Charron. Mek2 is dispensable for mouse growth and development. Molecular and Cellular Biology, 23(14):4778–4787, 2003.
Marie A. Bogoyevitch and Naomi W. Court. Counting on mitogen-activated protein kinases - ERKs 3, 4, 5, 6, 7 and 8. Cellular Signalling, 16(12): 1345–1354, 2004.
Barbara Bonnesen, Cathrine Orskov, Susanne Rasmussen, Peter Johannes Holst, Jan Pravsgaard Christensen, Karsten Wessel Eriksen, Klaus Qvortrup, Niels Odum, and Tord Labuda. Mek kinase1 activity is required for definitive erythropoiesis in the mouse fetal liver. Blood, 106(10):3396–3404, 2005.
F. Bost, M. Aouadi, L. Caron, P. Even, N. Belmonte, M. Prot, C. Dani, P. Hofman, G. Pages, J. Pouyssegur, Y. Le Marchand-Brustel, and B. Binetruy. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes, 54(2):402–411, 2005.
Karen Brami-Cherrier, Emmanuel Valjent, Denis Herve, Joanne Darragh, Jean-Christophe Corvol, Christiane Pages, Arthur J. Simon, Jean-Antoine Girault, and Jocelyne Caboche. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. The Journal of Neuroscience, 25(49):11444–11454, 2005.
D. Brancho, N. Tanaka, A. Jaeschke, J.J. Ventura, N. Kelkar, Y. Tanaka, M. Kyuuma, T. Takeshita, R.A. Flavell, and R.J. Davis. Mechanism of p38 MAP kinase activation in vivo. Genes and Development, 17(16):1969–1978, 2003.
Deborah Brancho, Juan-Jose Ventura, Anja Jaeschke, Beth Doran, Richard A. Flavell, and Roger J. Davis. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Molecular and Cellular Biology, 25(9):3670–3681, 2005.
Julian C. Braz, Orlando F. Bueno, Qiangrong Liang, Benjamin J. Wilkins, Yan-Shan Dai, Stephanie Parsons, Joseph Braunwart, Betty J. Glascock, Raisa Klevitsky, Thomas F. Kimball, Timothy E. Hewett, and Jeffery D. Molkentin. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. Journal of Clinical Investigation, 111(10):1475–1486, 2003.
Melanie M. Brinkmann and Thomas F. Schulz. Regulation of intracellular signalling by the terminal membrane proteins of members of the Gammaherpsesviridae. Journal of General Virology, 87(5):1047–1074, 2006.
O. F. Bueno, L. J. De Windt, K. M. Tymitz, S. A. Witt, T. R. Kimball, R. Klevitsky, T. E. Hewett, S. P. Jones, D. J. Lefer, C. F. Peng, R. N. Kitsis, and J. D. Molkentin. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. The EMBO Journal, 19 (23):6341–6350, 2000.
Emilio Casanova, Sandra Fehsenfeld, Erich Greiner, Andrew Francis Stewart, and Guenther Schuetz. Construction of a conditional allele of RSK-B/MSK2 in the mouse. Genesis, 32:158–160, 2002.
A.P. Chen, M. Ohno, K.P. Giese, R. Kuhn, R.L. Chen, and A.j. Silva. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. Journal of Neuroscience Research, 83(1):28–38, 2006.
Hongbo Chi, Binfeng Lu, Mutsuhiro Takekawa, Roger J. Davis, and Richard A. Flavell. GADD45b/GADD45g and MEKK4 comprise a genetic pathway mediating STAT4-independent IFNg production in T cells. The EMBO Journal, 23(7):1576–1586, 2004.
Hongbo Chi, Matthew R. Sarkisian, Pasko Rakic, and Richard F. Flavell. Loss of mitogen-activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proceedings of the National Academy of Sciences of the USA, 102(10):3846–3851, 2005.
Ainsley A. Culbert, Stephen D. Skaper, David R. Howlett, Nicholas A. Evans, Laura Facci, Peter E. Soden, Zoe M. Seymour, Florence Guillot, Matthias Gaestel, and Jill C. Richardson. MAPKAP kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity: relevance to neuroinflammation in a transgenic mouse model of Alzheimer’s disease. Journal of Biological Chemistry, Epub ahead of print, 2006.
Roger J. Davis. Signal transduction by the JNK group of MAP kinases. Cell, 103(2):239–252, 2000.
Chen Dong, Derek D. Yang, Mark Wysk, Alan J. Whitmarsh, Roger J. Davis, and Richard A. Flavell. Defective T-cell differentiation in the absence of Jnk1. Science, 282(5396):2092–2095, 1998.
Chen Dong, Derek D. Yang, Cathy Tournier, Alan J. Whitmarsh, Jie Xu, Roger J. Davis, and Richard A. Flavell. JNK is required for effector T-cell function but not for T-cell activation. Nature, 405(6782):91–94, 2000.
Karin Ehrenreiter, Daniela Piazzolla, Vanishree Velamoor, Izabela Sobczak, J. Victor Small, Junji Takeda, Thomas Leung T, and Manuela Baccarini. Raf-1 regulates Rho signaling and cell migration. Genomics, 168(6):955–964, 2005.
J. Endo, N. Toyama-Sorimachi, C. Taya, S. Kuramochi-Miyagawa, K. Nagata, K. Kuida, T. Takashi, H. Yonekawa, Y. Yoshizawa, N. Miyasaka, and H. Karasuyama. Deficiency of a STE20/PAK family kinase LOK leads to the acceleration of LFA-1 clustering and cell adhesion of activated lymphocytes. FEBS Letters, 468(2-3):234–238, 2000.
Susan M. Ferguson, Stefania Fasano, Pengwei Yang, Riccardo Brambilla, and Terry E. Robinson. Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology, Epub ahead of print, 2006.
Matthias Gaestel. Mapkap kinases - mks- two’s company, three’s a crowd. Nature Reviews Molecular Cell Biology, 7(2):120–30, 2006.
G. Galabova-Kovacs, D. Matzen, D. Piazzolla, K. Meissl, T. Plyushch, A.P. Chen, A. Silva, and M. Baccarini. Essential role of B-raf in ERK activation during extraembryonic development. Proceedings of the National Academy of Science of the USA, 103(5):1325–1330, 2006.
Soula Ganiatsas, Lia Kwee, Yuko Fujiwara, Andrew Perkins, Tohru Ikeda, Mark A. Labow, and Leonard l. Zon. SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proceedings of the National Academy of Sciences, 95(12): 6881–6886, 1998.
T.P. Garrington, T. Ishizuka, P.J. Papst, K. Chayama, S. Webb, T. Yujiri, W. Sun, S. Sather, D.M. Russell, S.B. Gibson, and G. Keller. MEKK2 gene disruption causes loss of cytokine production in response to IgE and c-Kit ligand stimulation of ES cell-derived mast cells. The EMBO Journal, 20(19): 5387–5395, 2000.
S. Giroux, M. Tremblay, D. Bernard, J.F. Cardin-Girard, S. Aubry, L. Larouche, S. Rousseau, J. Huot, J. Landry, L. Jeannotte, and J. Charron. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Current Biology, 9(7): 369–372, 1999.
X. Gong, X. Wang, J. Han, I. Niesman, Q. Huang, and J. Horwitz. Development of cataractous macrophthalmia in mice expressing an active MEK1 in the lens. Investigative Ophthalmology and Visual Science, 42(3): 539–548, 2001.
Basuki K. Gunawan, Zhang-Xu Liu, Derick Han, Naoko Hanawa, William A. Gaarde, and Neil Kaplowitz. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Journal of Gastroenterology, 131 (1):165–178, 2006.
Zijian Guo, Gavin Clydesdale, Jinke Cheng, Kihwan Kim, Lin Gan, David J. McConkey, Stephen E. Ullricht, Yuan Zhuang, and Bing Su. Disruption of Mekk2 in mice reveals an unexpected role for MEKK2 in modulating T-cell receptor signal transduction. Molecular and Cellular Biology, 22(16): 5761–5768, 2002.
Zuoning Han, David L. Boyle, Lufen Chang, Brydon Bennett, Michael Karin, Li Yang, Anthony M. Manning, and Gary S. Firestein. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. The Journal of Clinical Investigation, 108 (1):73–81, 2001.
Ian S. Harris, Shaosong Zhang, Ilya Treskov, Attila Kovacs, Carla Weinheimer, and Anthony J. Muslin. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation, 110(6):718–723, 2004.
N. Hatano, Y. Mori, M. Oh-hora, A. Kosugi, T. Fujikawa, N. Nakai, H. Niwa, J. Miyazaki, and T. Hamaoka M. Ogata. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes to Cells, 8 (11):847–856, 2003.
Martin Hegen, Matthias Gaestel, Cheryl L. Nickerson-Nutter, Lih-Ling Lin, and Jean-Baptiste Telliez. MAPKAP kinase 2-deficient mice are restistant to collagen-induced arthritis. The Journal of Immunology, 177(3):1913–1917, 2006.
Lorin M. Henrich, Jeffrey A. Smith, Danielle Kitt, Timothy M. Errington, Binh Nguyen, Abdulmaged M. Traish, and Deborah A. Lannigan. Extracellular signal-regulated kinase 7, a regulator of hormone-dependent estrogen receptor destruction. Molecular and Cellular Biology, 23(17):5979–5988, 2003.
R. M. Hobbs, V. Silva-Vargas, R. Groves, and F. M.Watt. Expression of activated MEK1 in differentiating epidermal cells is sufficient to generate hyperproliferative and inflammatory skin lesions. The Journal of Investigative Dermatology, 123(3):503–515, 2004.
M. Hüser, J. Luckett, A. Chiloeches, K. Mercer, M. Iwobi S. Giblett, X.M. Sun, J. Brown, R. Marais, and C. Pritchard. MEK kinase activity is not necessary for Raf-1 function. The EMBO Journal, 20(8):1940–1951, 2001.
Carlo Iavarone, Mario Acunzo, Francesca Carlomagno, Annuziata Catania, Rosa M. Melillo, Stella M. Carlomagno, Massimo Santoro, and Mario Chiariello. Activation of the ERK8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutively active form of the proto-oncogene. The Journal of Biological Chemistry, 281(15):10567–10576, 2006.
Masamichi Imajo, Yoshiki Tsuchiya, and Eisuke Nishida. Regulatory mechanisms and functions of MAP kinase signaling pathways. IUBMB Life, 58 (5-6):312–317, 2006.
Yasuhiro Izumi, Shokei Kim, Minoru Yoshiyama, Yasuhiro Izumiya, Kaoru Yoshida, Atsushi Matsuzawa, Hidenori Koyama, Yoshiki Nishizawa, Hidenori Ichijo, and Hiroshi Iwao Yoshikawa J. Activation of apoptosis signal-regulating kinase 1 in injured artery and its critical role in neointimal hyperplasia. Circulation, 108(22):2812–2818, 2003.
Yasukatsu Izumi, Shokei Kim-Mistuyama, Minoru Yoshiyama, Takashi Omura, Masayuki Shiota, Atsushi Matsuzawa, Tokihito Yukimura, Toyoaki Murohara, Motohiro Takeya, Hidenori Ichijo, Junichi Yoshikawa, and Hiroshi Iwao. Important role of apoptosis signal-regulating kinase 1 in ischemia-induced angiogenesis. Atheriosclerosis, Thrombosis and Vascular Biology, 25(9):1877–83, 2005.
Y. Izumiya, S. Kim, Y. Izumi, K. Yoshida, M. Yoshiyama, A. Matsuzawa, H. Ichijo, and H. Iwao. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodelling. Circulation Research, 93(9):874–883, 2003.
Robert A. Kaiser, Orlando F. Bueno, Daniel J. Lips, Pieter A. Doevendans, Fred Jones, Thomas F. Kimball, and Jeffery D. Molkentin. Targeted inhibition of p38 mitogen-activated protein kinase antagonizes cardiac injury and cell death following ischemia-reperfusion in vivo. The Journal of Biological Chemistry, 279(15):15524–15530, 2004.
Tamihiro Kamata, Catrin A. Pritchard, and Andrew D. Leavitt. Raf-1 is not required for megakaryocytopoiesis or TPO-induced ERK phosphorylation. Blood, 103(7):2568–2570, 2004.
K. Kesavan, K. Lobel-Rice, W. Sun, R. Lapadat, S. Webb, G.L. Johnson, and T.P. Garrington. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. Journal of Cellular Physiology, 199 (1):140–148, 2004.
J.A. Knauf, X. Ma, E.P. Smith, L. Zhang, N. Mitsutake, X.H. Liao, S. Refetoff, Y.E. Nikiforov, and J.A. Fagin. Targeted expression of BRAFV 600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Research, 65(10):4238–4245, 2005.
M. Kohno and J. Pouyssegur. Targeting the ERK signaling pathway in cancer therapy. Annals of Medicine, 38(3):200–211, 2006.
Monika Kortenjann, Michael Nehls, Andrew J. H. Smith, Rita Carsetti, Julia Schueler, Gabriele Koehler, and Thomas Boehm. Abnormal bone marrow stroma in mice deficient for nemo-like kinase, nlk. European Journal of Immunology, 31(12):3580–3587, 2001a.
Monika Kortenjann, Michael Nehls, Andrew J.H. Smith, Rita Carsetti, Julia Schueler, Gabriele Koehler, and Thomas Boehm. Abnormal bone marrow stroma in mice deficient for nemo-like kinase, Nlk. European Journal of Immunology, 31(12):3580–3587, 2001b.
Alexey Kotlyarov, Armin Neininger, Carola Schubert, Rolf Eckert, Carmen Birchmeier, Hans-Dieter Volk, and Matthias Gaestel. MAPKAP kinase 2 is essential for LPS-induced TNF-a biosynthesis. Nature Cell Biology, 1(2): 94–97, 1999.
Chia-Yi Kuan, Derek D. Yang, Deborah R. Samanta Roy, Roger J. Davis, Pasko Rakic, and Richard A. Flavel. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron, 22(4):667–676, 1999.
Li-Fu Li, Lunyin Yu, and Deborah A. Quinn. Ventilation-induced neutrophil infiltration depends on c-Jun N-terminal kinase. American Journal of Respiratory and Critical Care Medicine, 169(4):518–524, 2004.
Xiaofan Li and Audrey Minden. Targeted disruption of the gene for the PAK5 kinase in mice. Molecular and Cellular Biology, 23(20):7134–7142, 2003.
Y. Li, T. Minamino, O. Tsukamoto, T. Yujiri, Y. Shintani, K. Okada, Y. Nagamachi, M. Fujita, A. Hirata, S. Sanada, H. Asanuma, S. Takashima, M. Hori, G.L. Johnson, and M. Kitakaze. Ablation of MEK kinase 1 suppresses intimal hyperplasia by impairing smooth muscle cell migration and urokinase plasminogen activator expression in a mouse blood-flow cessation model. Circulation, 111(13):1672–1678, 2005.
Hong-Hsing Liu, Min Xie, Michael D. Schneider, and Zhijian J Chen. Essential role of TAK1 in thymocyte development and activation. Proceedings of National Academy of Sciences of the USA, 103(31):11677–11682, 2006.
H.T. Lu, D.D. Yang, M. Wysk, E. Gatti, I. Mellman, R.J. Davis, and R.A. Flavell. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. The EMBO Journal, 18(7):1845–1857, 1999.
Stephan Ludwig, Oliver Planz, Stephan Pleschka, and Thorsten Wolff. Influenza-virus-induced signalling cascades: targets for antiviral therapy? Trends in Molecular Medicine, 9(2):46–52, 2003.
Stephan Ludwig, Stephan Pleschka, Oliver Planz, and Thorstein Wolff. Ringing the alarm bells: signalling and apoptosis in influenza virus infected cells. Cellular Microbiology, 8(3):375–386, 2006.
Masumi Maruyama, Yuki Yagasaki, Tatsuhiko Sudo, and Hiroyuki Osas. Renal abnormalities in mice caused by insufficiency of p38alpha. Journal of Receptors and Signal Transduction, 23(2-3):17–183, 2003.
A. Matsuzawa, H. Nishitoh, K. Tobiume, K Takeda, and H. Ichijo. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxidants and Redox Signaling, 4(3):415–425, 2002.
C. Mazzucchelli, C. Vantaggiato, A. Ciamei, S. Fasano, P. Pakhotin, W. Krezel, H. Welzl, D.P. Wolfer, G. Pages, O. Valverde, A. Marowsky, A. Porrazzo, P.C. Orban, , R. Maldonado, M.U. Ehrengruber, V. Cestari, H.P. Lipp, P.F. Chapman, J. Pouyssegur, and R. Brambilla. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron, 34(5):807–820, 2002.
Jinsong Meng, Yanghong Meng, Amanda Hanna, Christopher Janus, and Zhengping Jia. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. The Journal of Neuroscience, 25(28): 6641–6650, 2005.
K. Mercer, S. Giblett, S. Green, D. Lloyd D, S.D. Dias, M. Lumb, R. Marais, and C. Pritchard. Expression of endogenous oncogeneic V 600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Research, 65(24):11493–11500, 2005a.
Kathryn Mercer, Susan Giblett, Anthony Oakden, Jane Brown, Richard Marais, and Cathrin Pritchard. A-Raf and Raf-1 work together to influence transient ERK phosphorylation and G1/S cell cycle progression. Oncogene, 24 (33):5207–5217, 2005b.
Theresa Mikalsen, Nancy Gerits, and Ugo Moens. Inhibitors of signal transduction protein kinases as targets for cancer therapy. Biotechnology Annual Review, 16:153–223, 2006.
M. Mikula, M. Schreiber, Z. Husak, L. Kucerova, J. Rüth, R. Wieser, K. Zatloukal, H. Beug, E.F. Wagner, and M. Baccarini. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. The EMBO Journal, 20(8):1952–1962, 2001.
M. Milella, Z. Estrov, S. M. Kornblau, B. Z. Carter, M. Konopleva, A. Tari, W. D. Schober, D. Harris, C. E. Leysath, G. Lopez-Berestein, Z. Huang, and M. Andreeff. Synergistic induction of apoptosis by simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute myelogenous leukemia. Blood, 99(9):3461–3464, 2002.
T. Minamino, T. Yujiri, N. Terada, G.E. Taffet, L.H. Michael, G.L. Johnson, and M.D. Schneider. MEKK1 is essential for cardiac hypertrophy and dysfunction induced by Gq. Proceedings of the National Academy of Sciences of the USA, 99(2):3866–3871, 2002.
Chantale I. Mourin and Jacques Huot. Recent advances in stress signaling in cancer. Cancer Research, 64(5):1893–1898, 2004.
John S. Mudgett, Jixiang Ding, Lucia Guh-Siesel, Nicole A. Chartrain, Lu Yang, Shobhna Gopal, and Michael M. Chen. Essential role for p38a mitogen-activated protein kinase in placental angiogenesis. Proceedings of the National Academy of Sciences, 97(19):10454–10459, 2000.
S. Münter, M. Way, and F. Frischknecht. Signaling during pathogen infection (full text available 160506). STKE, 335, 2006.
Tanya Nekrasova, Carey Shive, Yuehua Gao, Kazuyuki Kawamura, Rocio Guardia, Gary Landreth, and Thomas G. Forsthuber. ERK1-deficient mice show normal T cell effector function and are highly susceptible to experimental autoimmune encephalomyelitis. The Journal of Immunology, 175 (4):2374–2380, 2005.
Kazuhiko Nishida, Osamu Yamaguchi, Shinichi Hirotani, Shungo Hikoso, Yoshiharu Higuchi, Tetsuya Watanabe, Toshihiro Takeda, Soh Osuka, Takashi Morita, Gen Kondoh, Yoshihiro Uno, Kazunori Kashiwase, Masayuki Taniike, Atsuko Nakai, Yasushi Matsumura, Kenneth R. Chien, Junji Takeda, Masatsugu Hori, and Kinya Otsu. p38a mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Molecular and Cellular Biology, 24(24): 10611–10620, 2004.
H. Nishina, C. Vaz, P. Bilia, M. Nghiem, I. Sasaki, J.L. De la Pompa, K. Furlonger, C. Paige, C. Hui, K.D. Fischer, H. Kishimoto, I. Iwatsubo, T. Katada, J.R. Woodgett, and J.M. Penninger. Defective liver formation and liver cell apoptosis in mice lacking the stress signaling kinase SEK/MKK4. Development, 126(3):505–516, 1999.
H. Nishitoh, A. Matsuzawa, K. Tobiume, K. Seagusa, K. Takeda, K. Inoue, S. Hori, A. Kakizuka, and H. Ichijo. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes and Development, 16(11):1345–1355, 2002.
Deborah Veis Novack, Li Yin, Amanda Hagen-Stapleton, Robert D. Schreiber, David V. Goeddel, F. Patrick Ross, and Steven L. Teitelbaum. The IkB function of NF-kB2 p100 controls stimulated osteoclastogenesis. Journal of Experimental Medicine, 198(5):771–781, 2003.
Martin Olivier, David J Gregory, and Geneviève Forget. Subversion mechanisms by which Leishmana parasites can escape the host immune response: a signalling point of view. Clinical Microbiology Reviews, 18(2): 293–305, 2005.
Veronika Olschläger, Stephan Pleschka, Timo Fischer, Hanns-Joachim Rziha, Walter Wurzer, Lothar Stitz, Ulf R. Rapp, Stephan Ludwig, and Oliver Planz. Lung-specific expression of active Raf kinase results in increased mortality of influenza a virus-infected mice. Oncogene, 23(39):6639–6646, 2004.
Emily Omori, Kunihiro Matsumoto, Hideki Sanjo, Shintaro Sato, Shizuo Akira, Robert C. Smart, and Jun Ninomiya-Tsuji. TAK1 is a master regulator of epidermal homeostasis involving skin inflammation and apoptosis. The Journal of Biological Chemistry, 281(28):19610–19617, 2006.
Kinya Otsu, Nobushige Yamashita, Kazuhiko Nishida, Shinichi, Hirotani, Osamu Yamaguchi, Tetsuya Watanabe, Shungo Hikoso, Yoshiharu Higuchi, Yasushi Matsumura, Masumi Maruyama, Tatsuhiko Sudo, Hiroyuki Osada, and Masatsugu Hori. Disruption of a single copy of the p38a MAP kinase gene leads to cardioprotection against ischemia-reperfusion. Biochemical and Biophysical Reserach Communications, 302(1):56–60, 2003.
Gille Pages, Sandrine Guerin, Dominique Grall, Frederic Bonino, Austin Smith, Fabienne Anjuere, Patrick Auberger, and Jacque Pouyssegur. Defective thymocyte maturation in p44MAP kinase (Erk1) knockout mice. Science, 286 (5443):1374–1377, 1999.
C.A. Pritchard, L. Bolin, R. Slattery, R. Murray R, and M. McMahon. Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-raf protein kinase gene. Current Biology, 6(5): 614–617, 1996.
Jian Qu, Xiaofan Li, Bennet G. Novitch, Ye Zheng, Matthew Kohn, Jian-Ming Xie, Spencer Kozinn, Roderick Bronson, Amer A. Beg, and Audrey Minden. PAK4 kinase is essential for embryonic viability and for proper neuronal development. Molecular and Cellular Biology, 23(20):7122–7133, 2003.
Jan H. Reiling, Kathrin T. Doepfner, Ernst Hafen, and Hugo Stocker. Diet-dependent effects of the Drosophila Mnk1/Mnk2 homolog Lk6 on growth via eIFeE. Current Biology, 15(1):24–30, 2005.
Jie Ren, Shaosong Zhang, Attila Kovacs, Yibin Wang, and Anthony J. Muslin. Role of p38a MAPK in cardiac apoptosis and remodeling after myocardial infarction. Journal of Molecular and Cellular Cardiology, 38(4): 617–623, 2005.
L. Revesz, E. Blum, F. E. Di Padova, T. Buhl, R. Feifel, H. Gram, P. Hiestand, U. Manning, and G. Rucklin. Novel p38 inhibitors with potent oral efficacy in several models of rheumatoid arthritis. Bioorganic and Medicinal Chemistry Letters, 14(13):3595–3599, 2004.
Mercedes Rincon, Herve Enslen, Joel Raingeaud, Michael Recht, Tyler Zapton, Michael S-S. Su, Laurie A. Penix, Roger J. Davis, and Richard A. Flavell. Interferon-g expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. The EMBO Journal, 17(10):2817–2829, 1998a.
Mercedes Rincon, Alan Whitmarsh, Derek D. Yang, Linda Weiss, Benoit Derijard, Prash Jayaraj, Roger J. Davis, and Richard A. Flavell. The JNK pathway regulates the in vivo deletion of immature CD4+CD8+ thymocytes. Journal of Experimental Medicine, 188(10):1817–1830, 1998b.
Philippe P. Roux and John Blenis. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiology and Molecular Biology Reviews, 68(2):320–344, 2004.
M. K. Saba-El-Leil1, F. D. J. Vella, B. Vernay, L. Voisin, L. Chen, N. Labrecque, S.-L. Ang, and S. Meloche. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO reports, 4(10):964–968, 2003.
Kanaga Sabapathy, Yinling Hu, Tuula Kallunki, Martin Schreiber, Jean-Pierre David, Wolfram Jochum, Erwin F. Wagner, and Michael Karin. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Current Biology, 9(3):116–125, 1999a.
Kanaga Sabapathy, Wolfram Jochum, Konrad Hochedlinger, Lufen Chang, Michael Karin, and Erwin F. Wagner. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mechanisms of Development, 89(1-2):115–124, 1999b.
Guadalupe Sabio, James Simon, Campbell Arthur, Yvonne Kuma, Mark Peggie, Julia Carr, Vicky Murray-Tait, Francisco Centeno, Michel Goedert, Nicholas A. Morrice, and Ana Cuenda. p38g regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. The EMBO Journal, 24(16):1134–1145, 2005.
J. Sadoshima, O. Montagne, Q. Wang, G. Yang, J. Warden, J. Liu, G. Takagi, V. Karoor, C. Hong, G.L. Johnson, D.E. Vatner, and S.F. Vatner. The MEKK1-JNK pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy. Journal of Clinical Investigations, 110(2): 271–279, 2002.
Matthew P. Saelzler, Christy C. Spackman, Yuru Liu, Lesly C. Martinez, Jeremy P. Harris, and Mark K. Abe. ERK8 down-regulates transactivation of the glucocorticoid receptor through Hic-5. The Journal of Biological Chemistry, 281(24):16821–16832, 2006.
Toshiharu Sakurai, Shin Maeda, Lufen Chang, and Michael Karin. Loss of hepatic NF-kB activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proceedings of the National Academy of Sciences of the USA, 103(28):10544–10551, 2006.
Shintaro Sato, Hideki Sanjo, Kiyoshi Takeda, Jun Ninomiya-Tsuji, Masahiro Yamamoto, Taro Kawai, Kunihiro Matsumoto, Osamu Takeuchi, and Shizuo Akira. Essential function for the kinase TAK1 in innate and adaptive immune response. Nature Immunology, 6(11):1087–1095, 2005.
Koji Sayama, Yasushi Hanakawa, Hiroshi Nagai, Yuji Shirakata, Xiuju Dai, Satoshi Hirakawa, Sho Tokumaru, Mikiko Tohyama, Lujun Yang, Shintaro Sato, Akira Shizuo, and Koji Hashimoto. TAK1 is essential for differentiation and the prevention of apoptosis in epidermis. Journal of Biological Chemistry, 281(31):22013–22020, 2006.
Jörn M. Schattenberg, Rajat Singh, Yongjun Wang, Jay H. Lefkowitch, Raina M. Rigoli, Philipp E. Scherer, and Mark J. Czaja. JNK1 but not JNK2 promotes the development of steatoheptatis in mice. Hepatology, 43(1): 163–172, 2006.
Stefanie Schumacher, Kathrin Laas, Shashi Kant, Yu Shi, Alexey Kotlyarov, and Matthias Gaestel. Scaffolding by ERK3 regulates MK5 in development. The EMBO Journal, 23(24):1–10, 2004.
Michael Schumann and Matthias Dobbelstein. Adenovirus-induced extracellular signal-regulated kinase phosphorylation during the late phase of infection enhances viral protein levels and virus progeny. Cancer Research, 66 (3):1282–1288, 2006.
J. S. Sebolt-Leopold and R. Herrera. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature Reviews Cancer, 4(12):937–947, 2004.
J.C. Selcher, T. Nekrasova, R. Paylor, G.E. Landreth, and J.D. Sweatt. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learning and Memory, 8(1):11–19, 2001.
Ole Morten Seternes, Theresa Mikalsen, Bjarne Johansen, Espen Michaelsen, Chris G. Armstrong, Nick A. Morrice, Benjamin Turgeon, Sylvain Meloche, Ugo Moens, and Stephen M. Keyse. Activation of MK5/PRAK by the atypcial MAP kinase ERK3 defines a novel signal transduction pathway. The EMBO Journal, 23(24):4780–4791, 2004.
Yu Shi, Alexey Kotlyarov, Kathrin Laas, Achim D. Gruber, Elke Butt, Katrin Marcus, Helmut E. Meyer, Anke Friedrich, Hans-Dieter Volk, and Matthias Gaestel. Elimination of protein kinases MK5/PRAK activity by targeted homologous recombination. Molecular and Cellular Biology, 23(21): 7732–7741, 2003.
Jae-Hyuck Shim, Changchun Xian, Amber E. Paschal, Shannon T. Bailey, Ping Rao, Matthew S. Hayden, Ki-Young Lee, Crystal Bussey, Michael Steckel, Nobuyuki Tanaka, Gen Yamada, Shizuo Akira, Kunihiro Matsumoto, and Sankar Gosh. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes and Development, 19(22): 2668–2681, 2005.
Reiko Shinkura, Kazuhiro Kitada, Fumihiko Matsuda, Kei Tashiro, Koichi Ikuta, Misao Suzuki, Katsumi Kogishi, Tadao Serikawa, and Tasuku Honjo. Alymphoplasia is caused by a point mutation in the mouse gene encoding NF-k b-inducing kinase. Nature Genetics, 22(1):74–77, 1999.
Keisuke Shiroto, Hajime Otani, Fumio Yamamoto, Chi-Kuang Huang, Nilanjana Maulik, and Dipak K. Das. MK2-∕- gene knockout mouse hearts carry anti-apoptotic signal and are resistant to ischemia reperfusion injury. Journal of Molecular and Cellular Cardiology, 38(1):93–97, 2005.
Vadim V. Sumbayev and Inna M. Yasinska. Role of MAP kinase-dependent apoptotic pathway in innate immune responses and viral infections. Scandinavian Journal of Immunology, 63(6):391–400, 2006.
Hideo Tachibana, Cinzia Perrino, Hideyuki Takaoka, Roger J. Davis, Sathyamangla V. Naga Prasad, and Howard A. Rockman. JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochemical and Biophysical Research Communication, 343(4):1060–1066, 2006.
Yoko Takanami-Ohnishi, Shinya Amano, Sadao Kimura, Sachie Asada, Atsushi Utani, Masumi Maruyama, Hiroyuki Osada, Hajime Tsunoda, Yoko Irukayama-Tomobe, Katsutoshi Goto, Michael Karin, Tasuhiko Sudo, and Yoshitoshi Kasuya. Essential role of p38 mitogen-activated protein kinase in contact hypersensitivity. The Journal of Biological Chemistry, 277(40): 37896–37903, 2002.
Kumiko Tamura, Tatsuhiko Sudo, Uwe Senftleben, Agnes M. Dadak, Randall Johnson, and Michael Karin. Requirement for p38a in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell, 102(2):221–231, 2000.
N. Tanaka, M. Kamanaka, H. Enslen, C. Dong, M. Wysk, R.J. Davis, and R.A. Flavell. Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Reports, 3(8):785–791, 2002.
Nikos Tapinos and Anura Rambukkana. Insights into regulation of human Schwann cell proliferation by Erk1/2 via a MEK-independent and p56Lck-dependent pathway from leprosy bacilli. Proceedings of the National Academy of Sciences of the United States of America, 102(26):9188–9198, 2005.
Anne Barbara Tietz, Antje Malo, Joachim Diebold, Alexey Kotlyarov, Andreas Herbst, Frank T. Kolligs, Barbara Brandt-Nedelev, Walter Halangk, Matthias Gaestel, Burkhard Göke, and Claus Schäfer. Gene deletion of MK2 inhibits TNFa and IL-6 and protects against cerulein-induced pancreatitis. American Journal of Physiology Gastrointestinal and Liver Physiology, 290(6): G1298–306, 2006.
K. Tobiume, A. Matsuzawa, T. Takahashi, H. Nishitoh, K. Morita, K. Takeda, O. Minowa, K. Miyazono, T. Noda, and H. Ichijo. ASK1 is required for sustained activations of JNK/p38 map kinases and apoptosis. EMBO Reports, 21(31):222–228, 2001.
Gürol Tuncman, Jiro Hirosumi, Giovanni Solinas, Lufen Chang, Michael Karin, and Gökhan S. Hotamisligil. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proceedings of the National Academy of Sciences of the USA, 103(28):10741–10746, 2006.
Sophie Turban, Victoria A. Beardmore, Julia M. Carr, Kei Sakamoto, Eric Hajduch, J. Simon C. Arthur, and Harinder S. Hundal. Insulin-stimulated glucose uptake does not require p38 mitogen-activated protein kinase in adipose tissue or skeletal muscle. Diabetes, 54(11):3161–3168, 2005.
Takeshi Ueda, Rie Watanabe-Fukunaga, Hidehiro Fukuyama, Shigekazu Nagata, and Rikiro Fukunaga. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Molecular and Cellular Biology, 24(15):6539–6549, 2004.
Daniel Umbricht, Dimitri Vyssothky, Alexander Latanov, Roger Nitsch, Riccardo Brambiilla, Patrizia D’Adamo, and Hans-Peter Lipp. Midlatency auditory event-related potentials in mice: comparison to midlatency auditory ERPs in humans. Brain Research, 1019(1-2):189–200, 2004.
Teiji Wada and Josef M. Penninger. Mitogen-activated protein kinases in apoptosis regulation. Oncogene, 23(16):2838–2849, 2004.
Y. Wada, T. Nakajima-Yamada, K. Yamada, J. Tsuchida, T. Yasumoto, T. Shimozato, K. Aoki, T. Kimura, and S. Ushiyama. R-130823, a novel inhibitor of p38 MAPK, ameliorates hyperalgesia and swelling in arthritis models. European Journal of Pharmacology, 506(3):285–295, 2005.
R.A. Wang, H. Zhang, S. Balasenthil, D. Medina, and R. Kumar. PAK1 hyperactivation is sufficient for mammary gland tumor formation. Oncogene, 25:2931–2936, 2006a.
Rui-An Wang, Ratna K. Vadlamudi, Rozita Bagheri-Yarmand, Iwan Beuvink, Nancy E. Hynes, and Rakesh Kumar. Essential functions of p21-activated kinase 1 in morphogenesis and differentiation of mammary glands. Journal of Cell Biology, 161(3):583–592, 2003.
X. Wang and C. Tournier. Regulation of cellular functions by the ERK5 signalling pathway. Cellular Signalling, 18(6):753–760, 2006.
X. Wang, A.J. Merritt, J. Seyfried, C. Guo, E.S. Papadakis, K.G. Finegan, M. Kayahara, J. Dixon, R.P. Boot-Handford, E.J. Cartwright, U. Mayer, and C. Tournier. Targeted deletion of MEK5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Molecular and Cellular Biology, 25(1):336–345, 2005.
Xinkang Wang, Lin Xu, Hugh Wang, Peter R. Young, Matthias Gaestel, and Giora Z. Feuerstein. Mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 deficiency protects brain from ischemic injury in mice. The Journal of Biological Chemistry, 277(46):43968–43972, 2002.
Yongjun Wang, Rajat Singh, Jay H. Lefkowtich, Raina M. Rigoli, and Mark J. Czaja. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase 8 and the mitochondrial death pathway. The Journal of Biological Chemistry, 281(22):15258–15267, 2006b.
Giselle R. Wiggin, Ana Soloaga, Julia M. Foster, Victoria Murray-Tait, Philip Cohen, and J. Simon C. Arthur. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Molecular and Cellular Biology, 22(8):2871–2881, 2002.
L. Wojnowski, A.M. Zimmer, T.W. Beck, H. Hahn, R. Bernal, U.R. Rapp, and A. Zimmer. Endothelial apoptosis in Braf-deficient mice. Nature Genetics, 16(3):293–297, 1997.
L. Wojnowski, L.F. Stancato, A.M. Zimmer, H. Hahn, T.W. Beck, A.C. Larner, U.R. Rapp, and A. Zimmer. C-raf-1 protein kinase is essential for mouse development. Mechanisms of Development, 76(1-2):141–149, 1998.
Osamu Yamaguchi, Yoshiharu Higuchi, Shinichi Hirotani, Kazunori Kashiwase, Hiroyuki Nakayama, Shungo Hikoso, Toshihiro Takeda, Tetsuya Watanabe, Michio Asahi, Masayuki Taniike, Yasushi Matsumura, Ikuko Tsujimoto, Kenichi Hongo, Satoshi Kusakari, Kazuhiko Nishida, Hidenori Ichijo, Masatsugu Hori, and Kinya Otsu. Targeted deletion of apoptosis signal-regulating kinase1 attenuates left ventricular remodeling. Proceedings of the National Academy of Sciences of the USA, 100(26):15883–15888, 2003.
Osamu Yamaguchi, Tetsuya Watanabe, Kazuhiko Nishida, Kazunori Kashiwase, Yoshiharu Higuchi, Toshihiro Takeda, Shungo Hikoso, Shinichi Hirotani, Michio Asahi, Masayuki Taniike, Atsuko Nakai, Ikuko Tsujimoto, Yasushi Matsumura, Jun ichi Miyazaki, Kenneth R. Chien, Atsushi Matsuzawa, Chiharu Sadamitsu, Hidenori Ichijo, Manuela Baccarini, Masatsugu Hori, and Kinya Otsu. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. Journal of Clinical Investigation, 114(7):937–943, 2004.
S. Yamamoto, G. Yang, D. Zablocki, J. Liu, C. Hong, , S.J. Kim, S. Soler, M. Odashima, J. Thaisz, G. Yehia, C.A. Molina, A. Yatani, D.E. Vatner, S.F. Vatner, and J. Sadoshima. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. Journal of Clinical Investigation, 111(10):1463–1474, 2003.
L. Yan, J. Carr, P.R. Ashby PR, V. Murry-Tait, C. Thompson, and J.S. Arthur. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Developmental Biology, 3(11):1–21, 2003.
D. Yang, C. Tournier, M. Wysk, H.T. Lu, J. Xu, R.J. Davis, and R.A. Flavell. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proceedings of the National Academy of Science of the USA, 94(7):3004–3009, 1997a.
Derek D. Yang, Chia-Yi Kuan, Alan J. Whitmarsh, Mercedes Rincon, Timothy S. Zheng, Roger J. Davis, Pasko Rakic, and Richard A. Flavell. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature, 389(6653):865–870, 1997b.
Derek D. Yang, Dietrich Conze, Alan J. Whitmarsh, Tamera Barrett, Roger J. Davis, Mercedes Rincon, and Richard A. Flavell. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity, 9(4): 575–585, 1998.
J. Yang, Y. Lin, Z. Guo, J. Cheng, J. Huang, W. Liao L. Deng, Z. Chen, Z. Liu, and B. Su. The essential role of MEKK3 in TNF-induced NF-kB activation. Nature Immunology, 2(7):620–624, 2001.
Jianhua Yang, Melynda Boerm, Marya McCarty, Corazon Bucana, Isaiah J. Fidler, Yuan Zhuang, and Bing Su. Mekk3 is essential for early embryonic cardiovascular development. Nature Genetics, 24:309–313, 2000.
Y. Yao, W. Li, J. Wu, U.A. Germann, M.S.S. Su, K. Kuida, and D. M. Boucher. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proceedings of the National Academy of Sciences of the USA, 100(22):12759–12764, 2003.
Li Yin, Lin Wu, Holger Wesche, Cora D. Arthur, J. Michael White, David V. Goeddel, and Robert D. Schreiber. Defective lymphotoxin-b receptor-induced NFkB transcriptional activity in NIK-deficient mice. Science, 291(5511):2162–2165, 2001.
T. Yujiri, S. Sather, G.R. Fanger, and G.L. Johnson. Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted disruption. Science, 282(5395):1911–1914, 1998.
T. Yujiri, M. Ware, C. Widmann, R. Oyer, D. Russell, E. Chan, Y. Zaitsu, P. Clarke, K. Tyler, Y. Oka, G.R. Fanger, P. Henson, and G.L. Johnson. MEK kinase 1 gene disruption alters cell migration and c-Jun NH_2-terminal kinase regulation but does not cause a measurable defect in NF-kB activation. Proceedings of the National Academy of Sciences of the USA, 97(13): 7272–7277, 2000.
D. Zhang, V. Gaussin, G.E. Taffet, N.S. Belaguli, M. Yamada, R.J. Schwartz, L.H. Michael, P.A. Overbeek, and M.D. Schneider. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nature Medicine, 6(5):556–563, 2000.
Lin Zhang, Wei Wang, Yasuhito Hayashi, James V. Jester, David E. Birk, Min Gao, Chia-Yang Liu, Winston W.-Y. Kao, Michael Karin, and Ying Xia. A role for MEK kinase 1 in TGF-b/activin-induced epithelium movement and embryonic eyelid closure. The EMBO Journal, 22(17):4443–4454, 2003a.
R. Zhang, S. Murakami, F. Coustry, Y. Wang, and B. de Crombrugghe. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proceedings of the National Academy of Sciences of the USA, 103(2):365–370, 2006.
Shaosong Zhang, Jie Ren, Cindy E. Zhang, Ilya Treskov, Yibin Wang, and Anthony J. Muslin. Role of 14-3-3 mediated p38 mitogen-activated protein kinase inhibition in cardiac myocyte survival. Circulation Research, 93(11): 1026–1028, 2003b.
Shaosong Zhang, Carla Weinheimer, Michael Courtois, Attila Kovacs, Cindy E. Zhang, Alec M. Cheng, Yibin Wang, and Anthony J. Muslin. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. Journal of Clinical Investigation, 111(6):833–841, 2003c.
1 Abbreviations: aa: Amino Acid, ASO’s: Antisense Oligonucleotides, ca: Constitutive Active, dn: Dominant Negative, DNFB: 2.5-Dinitro-1-Fluorobenzene, KI: Knock-in, KO: Knock-out, LV: Left Ventricular, MAP kinase: Mitogen Activated Protein Kinase, MAPKK: Mitogen Activated Protein Kinase Kinase, MAPKKK: Mitogen Activated Protein Kinase Kinase Kinase, MAPKAPK: Mitogen Activated Protein Kinase-Activated Protein Kinase, MIP-2: Macrophage Inflammatory Protein-2, MMP: Matrix Metalloproteinase, MPO: Myeloperoxidase, NTG: Non-Transgenic, TAC: Transverse Aortic Constriction, TG: Transgenic, VILI: Ventilator Induced Lung Injury, WT: Wild-Type .